B 8224:2016
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
目 次
ページ
序文 ··································································································································· 1
1 適用範囲························································································································· 1
2 引用規格························································································································· 1
3 用語及び定義 ··················································································································· 1
4 共通事項························································································································· 1
4.1 通則 ···························································································································· 1
4.2 試料採取 ······················································································································ 1
4.3 分析方法 ······················································································································ 1
4.4 試験方法 ······················································································································ 2
4.5 定量範囲 ······················································································································ 2
4.6 繰返し精度 ··················································································································· 2
4.7 水 ······························································································································· 2
4.8 試薬 ···························································································································· 4
4.9 器具類 ························································································································· 4
4.10 検量線[吸光光度法,原子吸光法,フレーム光度法,ICP発光分光分析法,ICP質量分析法,イオ
ンクロマトグラフ法,イオン電極法,有機体炭素(TOC)分析法及び流れ分析法] ·························· 5
4.11 結果の表示 ·················································································································· 5
5 試料及び試料採取 ············································································································· 5
5.1 試料 ···························································································································· 5
5.2 試料採取 ······················································································································ 5
5.3 試料の取扱い ················································································································ 6
5.4 試料の保存処理 ············································································································· 7
5.5 試験時期 ······················································································································ 7
6 試料の前処理 ··················································································································· 7
6.1 一般事項 ······················································································································ 7
6.2 塩酸又は硝酸酸性で煮沸 ································································································· 7
6.3 塩酸又は硝酸による溶解 ································································································· 8
6.4 硝酸と過塩素酸とによる分解 ··························································································· 8
6.5 硝酸と硫酸とによる分解 ································································································· 9
6.6 フレーム原子吸光法,電気加熱原子吸光法,ICP発光分光分析法及びICP質量分析法を適用する場合
の前処理 ····························································································································· 9
7 pH································································································································ 10
7.1 一般事項 ····················································································································· 10
7.2 ガラス電極法 ··············································································································· 10
7.3 pHプロセス用分析装置による測定方法 ············································································· 13
B 8224:2016 目次
(2)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ページ
8 電気伝導率 ····················································································································· 14
8.1 一般事項 ····················································································································· 14
8.2 一般試験 ····················································································································· 14
8.3 酸電気伝導率 ··············································································································· 16
8.4 電気伝導率プロセス用分析装置による測定方法 ··································································· 18
9 酸消費量························································································································ 20
9.1 一般事項 ····················································································································· 20
9.2 酸消費量(pH 4.8) ······································································································· 20
9.3 酸消費量(pH 8.3) ······································································································· 21
10 硬度 ···························································································································· 22
10.1 一般事項 ···················································································································· 22
10.2 全硬度 ······················································································································· 22
10.3 カルシウム硬度 ··········································································································· 24
10.4 マグネシウム硬度 ········································································································ 24
11 蒸発残留物 ··················································································································· 25
11.1 一般事項 ···················································································································· 25
11.2 全蒸発残留物 ·············································································································· 25
11.3 溶解性蒸発残留物 ········································································································ 26
12 有機体炭素(TOC) ······································································································· 26
12.1 一般事項 ···················································································································· 26
12.2 燃焼酸化−赤外線式TOC分析法 ···················································································· 27
12.3 TOCプロセス用分析計(燃焼酸化−赤外線式)による測定方法 ··········································· 29
12.4 湿式酸化−赤外線式TOC分析法 ···················································································· 30
12.5 TOCプロセス用分析計(湿式酸化−赤外線式)による測定方法 ··········································· 32
13 ヘキサン抽出物質 ·········································································································· 32
13.1 一般事項 ···················································································································· 32
13.2 試料採取 ···················································································································· 33
13.3 抽出法 ······················································································································· 34
14 溶存酸素 ······················································································································ 36
14.1 一般事項 ···················································································································· 36
14.2 インジゴカルミン比色法 ······························································································· 36
14.3 隔膜電極法 ················································································································· 39
14.4 光学式センサ法 ··········································································································· 43
14.5 溶存酸素プロセス用分析装置による測定方法 ···································································· 45
15 塩化物イオン(Cl−) ····································································································· 47
15.1 一般事項 ···················································································································· 47
15.2 チオシアン酸水銀(II)吸光光度法 ················································································· 47
15.3 塩化銀比濁法 ·············································································································· 48
15.4 硝酸銀滴定法 ·············································································································· 49
B 8224:2016
(3)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ページ
15.5 イオン電極法 ·············································································································· 51
15.6 イオンクロマトグラフ法 ······························································································· 53
15.7 流れ分析法 ················································································································· 56
16 亜硫酸イオン(SO32−) ·································································································· 59
16.1 一般事項 ···················································································································· 59
16.2 よう素滴定法 ·············································································································· 59
17 硫酸イオン(SO42−) ····································································································· 63
17.1 一般事項 ···················································································································· 63
17.2 硫酸バリウム比濁法 ····································································································· 63
17.3 重量法 ······················································································································· 64
17.4 イオンクロマトグラフ法 ······························································································· 65
17.5 流れ分析法 ················································································································· 65
18 りん酸イオン(PO43−)及び加水分解性りん酸イオン ··························································· 68
18.1 一般事項 ···················································································································· 68
18.2 りん酸イオン ·············································································································· 69
18.3 加水分解性りん酸イオン ······························································································· 75
19 シリカ(SiO2) ············································································································· 76
19.1 一般事項 ···················································································································· 76
19.2 イオン状シリカ ··········································································································· 76
19.3 溶存及びコロイド状シリカ ···························································································· 84
19.4 全シリカ ···················································································································· 85
20 ヒドラジン(N2H4)[ヒドラジニウムイオン(N2H5+)] ························································ 87
20.1 一般事項 ···················································································································· 87
20.2 p-ジメチルアミノベンズアルデヒド吸光光度法 ·································································· 87
20.3 よう素滴定法 ·············································································································· 88
20.4 流れ分析法 ················································································································· 90
20.5 ヒドラジン(N2H4)[ヒドラジニウムイオン(N2H5+)]プロセス用分析装置(酸化還元電極)によ
る測定方法 ························································································································· 92
21 ナトリウム(Na) ········································································································· 93
21.1 一般事項 ···················································································································· 93
21.2 フレーム光度法 ··········································································································· 93
21.3 フレーム原子吸光法 ····································································································· 94
21.4 電気加熱原子吸光法 ····································································································· 95
21.5 ICP発光分光分析法 ····································································································· 96
21.6 ICP質量分析法 ··········································································································· 99
21.7 イオン電極法 ············································································································· 101
21.8 イオンクロマトグラフ法 ······························································································ 103
21.9 ナトリウム(Na)プロセス用分析装置(イオン電極法)による測定方法 ······························· 105
22 カルシウム(Ca) ········································································································ 106
B 8224:2016 目次
B 8224:2016 目次
(4)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ページ
22.1 一般事項 ··················································································································· 106
22.2 キレート滴定法 ·········································································································· 106
22.3 フレーム原子吸光法 ···································································································· 108
22.4 ICP発光分光分析法 ···································································································· 109
22.5 ICP質量分析法 ·········································································································· 109
22.6 イオンクロマトグラフ法 ······························································································ 111
23 マグネシウム(Mg) ····································································································· 111
23.1 一般事項 ··················································································································· 111
23.2 キレート滴定法 ·········································································································· 111
23.3 フレーム原子吸光法 ···································································································· 111
23.4 ICP発光分光分析法 ···································································································· 112
23.5 ICP質量分析法 ·········································································································· 113
23.6 イオンクロマトグラフ法 ······························································································ 113
24 銅(Cu) ···················································································································· 113
24.1 一般事項 ··················································································································· 113
24.2 ジエチルジチオカルバミド酸吸光光度法 ········································································· 113
24.3 クプリゾン吸光光度法 ································································································· 115
24.4 ジンコン吸光光度法 ···································································································· 116
24.5 フレーム原子吸光法 ···································································································· 117
24.6 電気加熱原子吸光法 ···································································································· 119
24.7 ICP発光分光分析法 ···································································································· 120
24.8 ICP質量分析法 ·········································································································· 123
25 亜鉛(Zn) ················································································································· 125
25.1 一般事項 ··················································································································· 125
25.2 フレーム原子吸光法 ···································································································· 125
25.3 電気加熱原子吸光法 ···································································································· 126
25.4 ICP発光分光分析法 ···································································································· 127
25.5 ICP質量分析法 ·········································································································· 127
26 鉄(Fe) ····················································································································· 127
26.1 一般事項 ··················································································································· 127
26.2 1,10-フェナントロリン吸光光度法 ·················································································· 127
26.3 2,4,6-トリ-2-ピリジル-1,3,5-トリアジン吸光光度法 ····························································· 129
26.4 フレーム原子吸光法 ···································································································· 131
26.5 電気加熱原子吸光法 ···································································································· 132
26.6 ICP発光分光分析法 ···································································································· 133
26.7 ICP質量分析法 ·········································································································· 133
附属書A(参考)試料及び試料採取 ······················································································· 139
B 8224:2016
(5)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ページ
附属書B(参考)濁度,アルカリ消費量,懸濁物質,100 ℃における過マンガン酸カリウムによる酸素消
費量,溶存酸素,残留塩素,塩化物イオン,硫酸イオン,りん酸イオン,アンモニア,ニッケル及びアル
ミニウムの測定に関する補足 ································································································ 152
附属書C(参考)脱ガス酸電気伝導率の測定 ··········································································· 200
附属書D(参考)腐食電位及び酸化還元電位の測定 ·································································· 204
参考文献 ··························································································································· 211
B 8224:2016 目次
B 8224:2016 目次
(6)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法第14条によって準用する第12条第1項の規定に基づき,一般社団法人火力
原子力発電技術協会(TENPES)及び一般財団法人日本規格協会(JSA)から,工業標準原案を具して日本
工業規格を改正すべきとの申出があり,日本工業標準調査会の審議を経て,経済産業大臣が改正した日本
工業規格である。
これによって,JIS B 8224:2005は改正され,この規格に置き換えられた。
この規格は,著作権法で保護対象となっている著作物である。
この規格の一部が,特許権,出願公開後の特許出願又は実用新案権に抵触する可能性があることに注意
を喚起する。経済産業大臣及び日本工業標準調査会は,このような特許権,出願公開後の特許出願及び実
用新案権に関わる確認について,責任はもたない。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
B 8224:2016
ボイラの給水及びボイラ水−試験方法
Boiler feed water and boiler water-Testing methods
序文
この規格は,1961年に制定され,その後5回の改正を経て今日に至っている。前回の改正は2005年に
行われたが,その後のJIS K 0102及びJIS B 8223の改正に対応するために改正した。
なお,対応国際規格は現時点で制定されていない。
1
適用範囲
この規格は,ボイラの給水(以下,給水という。),ボイラ水及び蒸気の試験方法について規定する。
なお,附属書Aに試料及び試料採取を,附属書Bに濁度,アルカリ消費量,懸濁物質,100 ℃における
過マンガン酸カリウムによる酸素消費量,溶存酸素,残留塩素,塩化物イオン,硫酸イオン,りん酸イオ
ン,アンモニア,ニッケル及びアルミニウムの測定に関する補足を,附属書Cに脱ガス酸電気伝導率の測
定を,附属書Dに腐食電位及び酸化還元電位の測定を,それぞれ参考として記載する。
2
引用規格
付表1に示す規格は,この規格に引用されることによって,この規格の規定の一部を構成する。これら
の引用規格は,その最新版(追補を含む。)を適用する。
3
用語及び定義
この規格で用いる主な用語及び定義は,JIS B 8223,JIS K 0101,JIS K 0102,JIS K 0211及びJIS K 0215
によるほか,次による。
3.1
プロセス用分析装置
プロセス用分析装置とは,プロセスにおいて,連続的に,又は一定周期ごとに分析する定置形の装置を
いう。
4
共通事項
4.1
通則
化学分析に共通する一般事項は,JIS K 0050による。
4.2
試料採取
試料採取に共通する一般事項は,JIS K 0094による。
4.3
分析方法
a) 吸光光度法 吸光光度法に共通する一般事項は,JIS K 0115による。吸収セルについて規定がない場
合には,光路長が10 mmのものを用いる。
2
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) 誘導結合プラズマ発光分光分析法 誘導結合プラズマ発光分光分析法(以下,“ICP発光分光分析法”
という。)に共通する一般事項は,JIS K 0116による。
c) 原子吸光法 原子吸光法には,フレーム原子吸光法,電気加熱方式原子吸光法(以下,“電気加熱原子
吸光法”という。)などがある。これらに共通する一般事項は,JIS K 0121による。
d) イオン電極法 イオン電極法に共通する一般事項は,JIS K 0122による。
e) イオンクロマトグラフ法 イオンクロマトグラフ法に共通する一般事項は,JIS K 0127による。
f)
高周波プラズマ質量分析法 高周波プラズマ質量分析法(以下,“ICP質量分析法”という。)に共通
する一般事項は,JIS K 0133による。
g) 流れ分析法 流れ分析法に共通する一般事項は,JIS K 0126による。
4.4
試験方法
この規格で規定していない試験方法は,JIS K 0101又はJIS K 0102による。
4.5
定量範囲
それぞれの試験方法に示してある定量範囲は,主として検量線の濃度(mg/L又はµg/L)で表示する。
4.6
繰返し精度
繰返し精度は,それぞれの試験方法の定量範囲内において繰返し試験で求めた変動係数(%)で示し,
次のとおりとする。
100
x
%
×
=σ
)
変動係数(
ここに,
σ: 標準偏差
x: 平均値
4.7
水
この規格で用いる水は,JIS K 0557で規定するA1〜A4の水とするが,各箇条で規定する場合には,そ
れに従う。
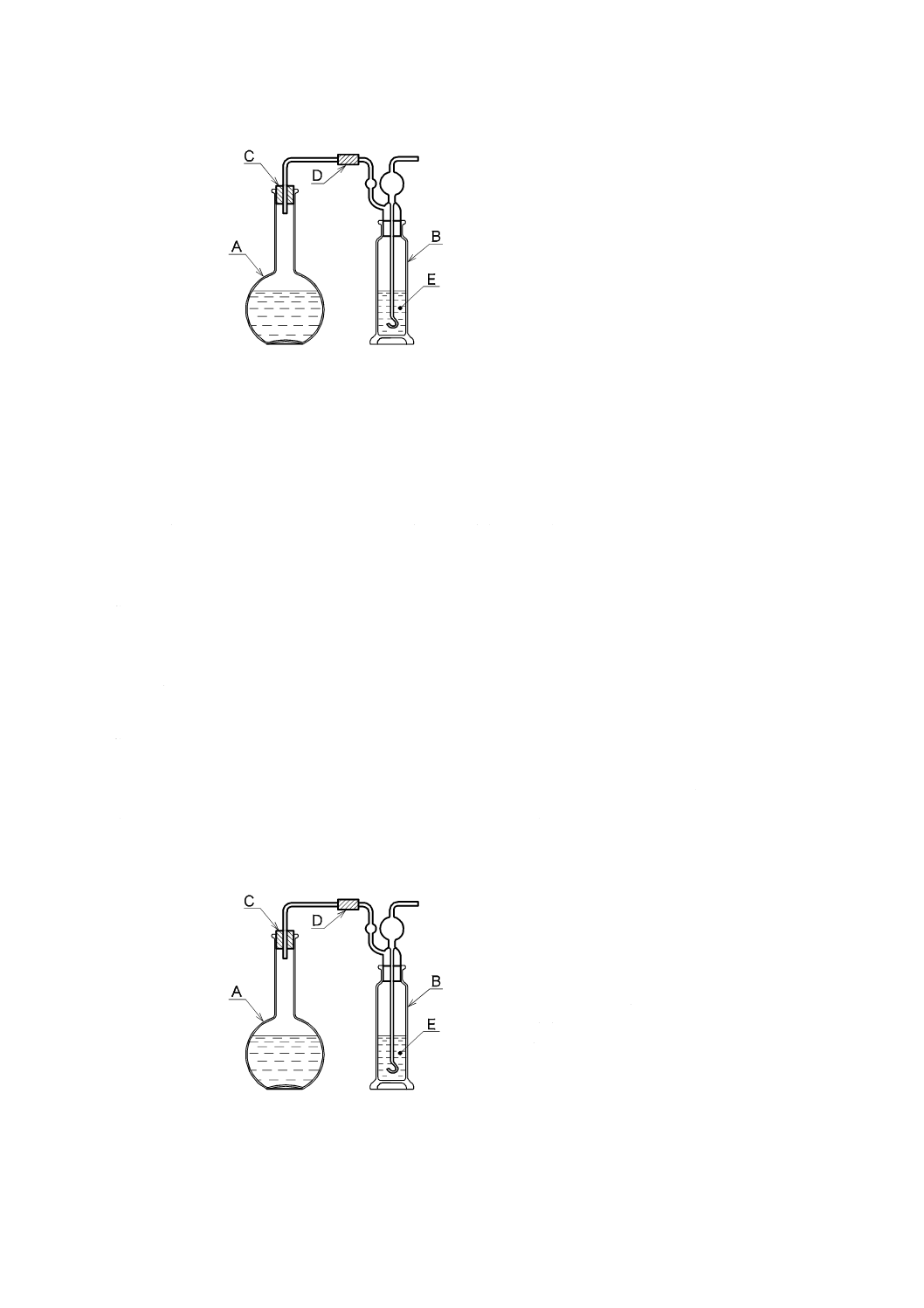
a) 二酸化炭素を含まない水[JIS K 0050の附属書E(特殊用途の水の調製方法及び保存方法)参照。]
二酸化炭素を除いた水の調製方法は,次の1)〜4) のいずれか,又はそれらの二つ以上を組み合わせ
たものを用い,使用時に調製する。保存する場合は,図1と同様な装置を用い,ガス洗浄瓶に水酸化
カリウム溶液(250 g/L)(JIS K 8574で規定する水酸化カリウムを用いて調製する。)又はJIS K 8603
で規定するソーダ石灰の二酸化炭素吸収用1号を入れ,空気中の二酸化炭素を遮断して保存する。
1) JIS K 0557で規定するA2又はA3の水をフラスコに入れ,約5分間煮沸して二酸化炭素を除去した
後,図1と同様な装置を用い,ガス洗浄瓶に水酸化カリウム溶液(250 g/L)又はJIS K 8603で規定
するソーダ石灰の二酸化炭素吸収用1号を入れ,空気中の二酸化炭素を遮断して放冷する。
2) JIS K 0557で規定するA2又はA3の水をフラスコに入れ,JIS K 1107で規定する窒素2級を約15
分間通気して,二酸化炭素を除去する。
3) JIS K 0557で規定するA2又はA3の水を,二酸化炭素分離膜を用いたガス分離管に通水し,二酸化
炭素を除去する。
4) JIS K 0557で規定するA2又はA3の水を,超音波振動装置で十分脱気を行い,二酸化炭素を除去す
る。
3
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A:平底フラスコ 1 000 mL
B:ガス洗浄瓶 250 mL
C:ゴム栓
D:ゴム管
E:水酸化カリウム溶液(250 g/L)
図1−二酸化炭素を含まない水の冷却,保存の一例
b) 溶存酸素を含まない水[JIS K 0050の附属書E(特殊用途の水の調製方法及び保存方法)参照。] 溶
存酸素を除いた水の調製方法は,次の1)〜5) のいずれか,又はそれらの二つ以上を組み合わせたもの
を用い,使用時に調製する。保存する場合は,図2のようにアルカリ性ピロガロール溶液を入れたガ
ス洗浄瓶を連結し,空気中の酸素を遮断して保存する。JIS K 8780で規定するピロガロール(1,2,3-
ベンゼントリオール)6 gを水50 mLに溶かし,着色瓶に保存する。別に,JIS K 8574で規定する水
酸化カリウム30 gを水50 mLに溶かす。使用時に両液を混合する。この溶液1 mLは,酸素約12 mL
(約17 mg)を吸収する。
1) JIS K 0557で規定するA2又はA3の水をフラスコに入れ,約5分間煮沸して溶存酸素を除去した後,
図2のようにアルカリ性ピロガロール溶液を入れたガス洗浄瓶を連結して,空気中の酸素を遮断し
て放冷する。
2) JIS K 0557で規定するA2又はA3の水をフラスコに入れ,JIS K 1107で規定する窒素2級を約15
分間通気して溶存酸素を除去する。
3) JIS K 0557で規定するA2又はA3の水を,酸素分離膜を用いたガス分離管に通水し,溶存酸素を除
去する。
4) JIS K 0557で規定するA2又はA3の水を,超音波振動装置で十分脱気を行い,溶存酸素を除去する。
5) JIS K 0557で規定するA2又はA3の精製直後の水を,JIS K 1107で規定する窒素2級を通じた三角
フラスコに泡立てないように採取したもの。
A:平底フラスコ 1 000 mL
B:ガス洗浄瓶 250 mL
C:ゴム栓
D:ゴム管
E:アルカリ性ピロガロール溶液
図2−溶存酸素を含まない水の冷却,保存の一例
4
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4.8
試薬
a) 試薬は,日本工業規格(以下,“JIS”という。)で規定するもので,試験に支障のないものを用いる。
JISで規定がない場合は,試験に支障がない品質のものを用いる。
電気加熱原子吸光法,ICP質量分析法など,ごく低濃度の試験には,特に高純度の試薬を用いる。
高純度の試薬には,JIS K 9901で規定する高純度試薬−硝酸,JIS K 9902で規定する高純度試薬−塩
酸,JIS K 9904で規定する高純度試薬−過塩素酸,JIS K 9905で規定する高純度試薬−硫酸などがあ
る。
滴定液類の標定には,JIS K 8005で規定する容量分析用標準物質を用いる。
b) 試薬類の濃度は,特に断らない限り,質量濃度はg/L又はmg/L,モル濃度はmol/L又はmmol/Lで示
す。
なお,化合物の質量は,名称の後に括弧で示し,無水物としての値を用いる。
標準液の濃度は,1 L中の質量(mg/L又はµg/L)で表す。
c) 試薬類の溶液名称の後に括弧で示す濃度は,標準液以外は概略の濃度であることを意味する。例えば,
水酸化ナトリウム溶液(0.1 mol/L)は,約0.10 mol/Lの水酸化ナトリウム溶液であることを示す。ま
た,液体試薬Aと液体試薬Bとの混合溶液の濃度は,A(a+b)で表す。この表し方はAとBとをa
mLとb mLとの割合で混合したことを示す。
なお,溶液名の前に示す濃度は,正確な濃度を意味する。端数のない数値で示し,別にファクタを
求めておく。
d) 試薬類の調製に用いる水は,4.7の水のうちのA3又はA4の水とするが,各箇条で規定する場合には,
それに従う。
e) 標準液を薄めて低濃度の標準液を調製する場合には,特に断りのない限り,10 mL以上の全量ピペッ
トでとる。
f)
試薬類の名称は,国際純正及び応用化学連合(IUPAC)の無機化学命名法及び有機化学命名法を基に
して,公益社団法人日本化学会が定めた化合物命名法及びJIS試薬の名称に整合させた。
g) 試薬類,廃液類などによる室内汚染,人体への吸収,付着などに注意する。また,その取扱いについ
ては,関連法令,規則などに従う。
h) 標準液は次による。
1) 標準液,混合標準液は,国家計量基準(計量法第134条)で規定するトレーサビリティが確保され
たものを用いる。
参考 トレーサビリティが確保された試薬としては,JCSSマークを付けたものがある。
2) 試験に用いる混合標準液は各試験方法の規定による。濃度保証された市販の分析用標準液を用いて
もよい。
3) 対象物質をそれぞれ単独で試験する場合には,必要な物質の標準液を調製する。
4.9
器具類
この規格で用いるガラス器具,磁器るつぼ,磁器蒸発皿,白金るつぼ,白金蒸発皿及びろ紙は,次によ
る。
なお,シリカ,ナトリウムを試験する場合には,ほうけい酸ガラスからのこれらの成分の溶出に十分に
注意する。
a) ガラス器具は,特に断らない限り,JIS R 3503及びJIS R 3505で規定するものを使用する。ただし,
特殊な器具を必要とする場合には,各箇条に,その一例を図示又は説明する。また,加熱操作を伴う
5
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
場合には,特に断らない限り,JIS R 3503で規定するほうけい酸ガラス−1を用いる。
デシケータに用いる乾燥剤は,特に断らない限り,JIS Z 0701で規定する包装用シリカゲル乾燥剤
A形1種を用いる。
b) 磁器るつぼ及び磁器蒸発皿は,それぞれJIS R 1301及びJIS R 1302で規定するものを使用する。
c) 白金るつぼ及び白金蒸発皿は,それぞれJIS H 6201及びJIS H 6202で規定するものを使用する。
d) ろ紙は,JIS P 3801で規定する定量分析用を使用する。ただし,ろ紙の種類は,各箇条で規定する。
4.10
検量線[吸光光度法,原子吸光法,フレーム光度法,ICP発光分光分析法,ICP質量分析法,イオ
ンクロマトグラフ法,イオン電極法,有機体炭素(TOC)分析法及び流れ分析法]
a) 検量線の作成に当たっては,試験方法に示す定量範囲内を4〜6段階に分け,これに一致するように標
準液をとる。
b) 検量線は定量範囲内について作成する。
c) 原子吸光法,フレーム光度法,ICP発光分光分析法,ICP質量分析法,イオンクロマトグラフ法,イ
オン電極法,有機体炭素(TOC)分析法及び流れ分析法の試験では,試験に際して新たに作成した検
量線を用い,多数の試料に対して同一の分析項目を連続して試験する場合には,試験の途中において,
適宜,標準液を用いて指示値(又はその比例値)の確認を行う。
d) 吸光光度法では,あらかじめ作成した検量線を用いることもできる。
4.11
結果の表示
試験結果の表示は,次による。
a) 試験方法が二つ以上あるときは,用いた試験方法を明記する。
b) 濃度はmg/L又はµg/Lで表す。
5
試料及び試料採取
5.1
試料
試料は,各種の試験を行うために給水,ボイラ水及び蒸気から採取した水を指し,試料は,それぞれの
採取位置における給水,ボイラ水及び蒸気を代表できるものでなければならない。
5.2
試料採取
試料採取は,次による。
5.2.1
試料容器
試料容器は,特に規定がない限り,JIS R 3503で規定するほうけい酸ガラス−1の無色の共栓瓶,JIS Z
1703で規定する共栓ポリエチレン瓶,共栓ポリプロピレン瓶などを用いる。また,試料容器は,試料に外
部からの物質の混入及び試料から試験成分が逃げるのを防ぐため,密栓できるものを用いる。ただし,栓
にはゴム,コルクなどを用いない。
未使用の新しい試料容器は,まず,温硝酸(1+10)又は温塩酸(1+10)で洗浄し,更に水又は中性洗
剤などを用いて洗浄する。洗浄後は水を満たしておく。
一度使用した試料容器を再使用する場合には,温塩酸(1+5)で洗浄し,更に水で十分に洗浄する。
5.2.2
試料採取位置
給水,ボイラ水及び蒸気の試料は,JIS B 8223で規定する位置,又は試験目的に最適な位置から採取す
る。
5.2.3
試料採取装置
給水,ボイラ水及び蒸気の試料を採取する場合には,試料の冷却部及び減圧系統を通して採取する。た
6
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
だし,試料が50 ℃未満,圧力2 MPa以下ならば冷却器及び減圧装置を省略することができる。
a) 試料導管 試料導管は,各系統から取り出した試料を冷却器に導くためのもので,その材料はJIS G
3463で規定するボイラ・熱交換器用ステンレス鋼鋼管,又はJIS G 3459で規定する配管用ステンレス
鋼鋼管を用いる。ただし,試験項目及び試料の濃度によっては,JIS G 3454で規定する圧力配管用炭
素鋼鋼管,又はJIS G 3456で規定する高温配管用炭素鋼鋼管を用いてもよい。
b) 冷却器 冷却器の材料は,高圧ボイラ用にはステンレス鋼を用いる。冷却器の能力は室温以下(溶存
酸素を定量する試料を採取する場合には,室温より約2 ℃低くする。)の試料が1 L/min以上の流量で
採取できるものとする。
c) 減圧装置 減圧装置は,試料採取系統の各弁を全開した状態で,試料が1〜2 L/minの割合で採取でき
るように減圧できるものとする。
d) ボイラ内試料採取管 ボイラは,試料が採取できる連続ブロー装置,ボイラ水試料採取用内管などを
備えたものとする。
e) 蒸気採取ノズル 蒸気採取ノズルは蒸気取入れ孔をもち,蒸気管にはめ合わせ,ノズルの蒸気取入れ
孔から流入した蒸気を試料導管に導き,採取できる構造とする。
5.2.4
試料採取操作
試料の採取操作は,次による。
a) 採取操作 ヘキサン抽出物質,溶存酸素,亜硫酸イオン及びヒドラジニウムイオンを試験する場合の
試料採取は,それぞれの試験項目による。これ以外の試験項目については,次による。
試料採取装置の試料出口管の先端に軟質塩化ビニル管を取り付け,流出する試料を試料容器の容量
の約1/4量を入れて激しく振り混ぜて洗浄(5回繰り返す)する。次に,試料容器の底面に軟質塩化
ビニル管の先端が接するようにして,試料容器の容量の約5倍量の試料を十分に流出させた後,軟質
塩化ビニル管を取り出し,共栓を試料で十分に洗った後,密栓する。
ただし,鉄,銅及び亜鉛を試験する場合には,軟質塩化ビニル管の表面に試料中の鉄,銅及び亜鉛
が吸着する可能性があるので,軟質塩化ビニル管が試料容器の内面及び試料容器中の試料液面に接し
ないように採取する。
b) 採取量 試料の採取量は,試験目的及び試験項目数によって決定する。
5.2.5
試料採取時の記録事項
試料採取時には,次の事項を記録する。その他必要に応じて追加する。
a) 試料の名称
b) 試料採取位置
c) 採取年月日及び時刻
5.2.6
留意事項
試料採取装置を新設した場合,又はボイラを長期間停止し,再始動する場合には,試料元弁を開いた後,
試料導管フラッシング弁を開き,十分に試料導管部を洗浄する。その後,化学分析系統だけに試料を流し
て,試料採取装置内の洗浄を行う。次に,水質監視計器を含めた全系統に試料を24時間以上流して全体の
洗浄を行う。
5.3
試料の取扱い
金属元素の試験は,試料中に含まれる全量について行う。このため試料に懸濁物がある場合は,十分に
振り混ぜて均一にした後,試料を採取して試験に用いる。ただし,金属元素のうちナトリウム,カルシウ
ム,マグネシウム及び陰イオンの試験では,特に断らない限り,ろ過した試料を用いる。
7
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
溶存状態のものだけを試験する場合には,試料採取後,直ちにろ紙5種C(又はろ紙6種)又は孔径0.45
〜1 µmのろ過材を用いてろ過し,初めのろ液約50 mLを捨て,その後のろ液を試料とする。
5.4
試料の保存処理
試験は,特に断らない限り,試料採取後,直ちに行う。直ちに試験ができずに保存(又は運搬が必要な
場合)する場合は,JIS K 0094の7.(試料の保存処理)に従って5.4.2のように行い,なるべく早く試験す
る。冷所に保存する場合には,凍結させないようにする。
5.4.1
試薬
試薬は,次による。
a) 塩酸 JIS K 8180で規定するもの。
b) 硝酸 JIS K 8541で規定するもの。
5.4.2
保存処理
保存処理は,次による。
a) 銅,亜鉛,鉄を試験する試料は,硝酸でpHを約1にする。鉄(II)を試験する試料は,塩酸を用いる。
電気加熱原子吸光法及びICP質量分析法を適用する場合は,JIS K 9901で規定する高純度試薬−硝酸
を用いる。
溶存状態の金属元素を試験する試料は,試料採取直後,5.3によって試料をろ過した後,硝酸を加え
てpHを約1にして保存する。
b) ヒドラジンの試験に用いる試料は,塩酸を加えてpHを2〜3にする。
c) ヘキサン抽出物質の試験に用いる試料は,塩酸(1+1)[5.4.1 a) の塩酸を用いて調製する。]を加え
てpHを約4にする。
d) 有機体炭素(TOC)の試験に用いる試料は,0〜10 ℃の暗所に保存し,できるだけ早く試験する。
5.5
試験時期
試験は,特に断らない限り,試料採取後,直ちに行う。保存処理を行うことによって,試料中の成分の
変質を防止できるものについては,5.4.2の操作を行った後,できるだけ早く試験する。
溶存酸素,亜硫酸イオンの試験は,試料採取現場において試料採取後,直ちに行う。pH,電気伝導率の
試験は,試料採取後,直ちに行う。緩衝作用の弱い試料のpHを測定する場合には,できるだけ空気との
接触を防いで流液形の測定セルを用いて測定する。
6
試料の前処理
6.1
一般事項
試料の前処理操作は,各試験項目で規定するが,金属元素の試験における前処理操作は,金属元素の種
類に関係なく共通するものがほとんどであるため,一括して6.2〜6.6で規定する。ただし,金属元素のう
ちナトリウム,カルシウム,マグネシウムなどの試験の前処理は,それぞれの試験項目において規定する。
金属元素の試験に用いる試料の前処理は,主として共存する有機物,懸濁物及び金属錯体の分解を目的
としている。前処理には,試料に各種の酸を加えて加熱する方法を用いるが,試料の状態及び試験の種類
によって適切な方法を選択する。
6.2
塩酸又は硝酸酸性で煮沸
この方法は,有機物及び懸濁物が極めて少ない試料及び溶存状態の金属元素を試験する場合に適用する。
6.2.1
試薬
試薬は,次による。
8
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) 塩酸 5.4.1 a) による。
b) 硝酸 5.4.1 b) による。
6.2.2
操作
操作は,次による。
a) 試料100 mLにつき塩酸5 mL又は硝酸5 mLを加える。溶存状態の金属元素を試験する場合には,5.3
によってろ過した試料を用いる。
b) 加熱して約10分間静かに煮沸する。
c) 放冷後,必要に応じて水で一定量にする。
6.3
塩酸又は硝酸による溶解
この方法は,有機物が少なく,懸濁物として水酸化物,酸化物,硫化物,りん酸塩などを含む試料に適
用する。
6.3.1
試薬
試薬は,次による。
a) 塩酸 5.4.1 a) による。
b) 硝酸 5.4.1 b) による。
6.3.2
操作
操作は,次による。
a) 試料を振り混ぜた後,直ちにビーカにとり,試料100 mLにつき塩酸5 mL又は硝酸5 mLを加える。
b) 加熱して液量が約15 mLになるまで濃縮する。
c) 不溶解物が残った場合には,ろ紙5種Bでろ過した後,水でよく洗浄する。
d) 放冷後,ろ液と洗液を適切な容量の全量フラスコに移し入れ,水を標線まで加える。
6.3.3
留意事項
塩酸と硝酸との混酸による溶解が有利な試料の場合には,6.3.2 b) までの操作を行った後,室温まで放
冷する。6.3.2 a) で塩酸を使用したときは硝酸5 mLを,硝酸を使用したときは塩酸5 mLを加え,時計皿
で覆い再び加熱し,激しい反応の終了後,時計皿を取り除き,更に加熱して窒素化合物を追い出し,約5 mL
になるまで濃縮する。この操作で酸が不足している場合には,適量の塩酸及び硝酸を加え,同じ操作で加
熱して溶かす。不溶解物が残った場合には,温水15 mLを加え6.3.2 c) 及び6.3.2 d) の操作を行う。
6.4
硝酸と過塩素酸とによる分解
この方法は,酸化されにくい有機物を含む試料に適用する。
6.4.1
試薬
試薬は,次による。
a) 過塩素酸 JIS K 8223で規定するもの。
b) 硝酸 5.4.1 b) による。
6.4.2
操作
操作は,次による。
a) 試料をよく振り混ぜ,直ちにその適量をビーカ又は磁器蒸発皿にとる。
b) 硝酸5〜10 mLを加え,加熱板上で静かに加熱して約10 mLになるまで濃縮し,放冷する。ケルダー
ルフラスコに移して分解してもよい。
c) 硝酸5 mLを加え,過塩素酸10 mLを少量ずつ加え,加熱を続け,過塩素酸の白煙が発生し始めたら,
時計皿で容器を覆い,過塩素酸が器壁を流下する状態に保って有機物を分解する。
9
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
d) 有機物が分解しないで残ったときは,更に硝酸5 mLを加えてc) の操作を繰り返す。
e) 放冷後,水を加えて液量を約50 mLに薄め,不溶解物が残った場合には,ろ紙5種Bを用いてろ過し,
水で洗い,ろ液と洗液を適切な容量の全量フラスコに移し入れ,水を標線まで加える。
警告 過塩素酸を用いる加熱分解操作は,試料の種類によっては爆発の危険性があるため,次のこ
とに注意する。
i)
酸化されやすい有機物は,過塩素酸を加える前に,b) の操作によって十分に分解してお
く。
ii) 過塩素酸の添加は,必ず濃縮液を放冷した後に行う。
iii) 必ず過塩素酸と硝酸とを共存させた状態で加熱分解を行う。
iv) 濃縮液を乾固させない。
6.5
硝酸と硫酸とによる分解
この方法は,多種類の試料に適用することができるが,水溶液をそのまま噴霧するフレーム原子吸光法
を適用する場合には,干渉があるので,注意する。
6.5.1
試薬
試薬は,次による。
a) 硝酸 5.4.1 b) による。
b) 硫酸(1+1) 水1容をビーカにとり,これを冷却し,かき混ぜながらJIS K 8951で規定する硫酸1
容を徐々に加える。
6.5.2
操作
操作は,次による。
a) 試料をよく振り混ぜ,直ちにその適量をビーカ又は磁器蒸発皿にとり,硝酸5〜10 mLを加える。
b) 加熱して,液量が約10 mLになったら,再び硝酸5 mLと硫酸(1+1)10 mLとを加え,硫酸の白煙
が発生し,有機物が分解するまで加熱する。ケルダールフラスコに移して分解してもよい。
c) 有機物の分解が困難なときは,更に硝酸10 mLを加え,b) の操作を繰り返す。
d) 放冷後,水で液量を約50 mLに薄める。不溶解物が残った場合には,ろ紙5種Bを用いてろ過し,水
で洗い,ろ液と洗液を適切な容量の全量フラスコに移し入れ,水を標線まで加える。
6.5.3
留意事項
鉛が含まれていて沈殿を生じる場合には,6.4又は次による。
6.5.2 b) の操作を行って溶液をほとんど蒸発乾固し,水約30 mLと塩酸15 mLとを加え,加熱して溶か
す。不溶解物がある場合には,ろ紙5種Bを用いてろ過した後,温塩酸(1+10)で洗浄する。放冷後,
ろ液と洗液とを適切な容量の全量フラスコに移し入れ,水を標線まで加える。
6.6
フレーム原子吸光法,電気加熱原子吸光法,ICP発光分光分析法及びICP質量分析法を適用する場
合の前処理
試料に含まれている有機物及び懸濁物の量,その存在状態及び適用しようとする原子吸光法,ICP発光
分光分析法,ICP質量分析法などの特徴を十分に考慮して6.2〜6.5に示した方法のうち最適なものを選択
して前処理する。
原子吸光法又はICP発光分光分析法に先立って溶媒抽出法を適用する場合の前処理は,特に断らない限
り,各箇条のとおりとし,妨害するおそれのある有機物その他の妨害物質を十分に分解する。
試料をそのまま噴霧する方法による原子吸光法又はICP発光分光分析法を適用する場合には,次に示す
前処理を行ってもよい。
10
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
有機物及び懸濁物が極めて少ない試料の場合は,6.2の操作を行う。有機物又は懸濁物を含む試料の一般
的な前処理方法としては,6.4又は6.5を適用する。この場合,白煙を十分に発生させて大部分の硫酸及び
過塩素酸を除去しておく。
ICP質量分析法の場合は,酸の種類及び濃度によって空試験値が無視できないことがあるので,測定す
る元素についてあらかじめ酸の種類及び濃度の影響について調べておく。
いずれの前処理方法を適用するかは,試料に一定量の目的成分を添加して回収試験を行い,その結果に
基づいて判断するとよい。
前処理定容した試料の酸の種類,濃度は,特に断らない限り,フレーム原子吸光法又はICP発光分光分
析法を適用する場合には,塩酸又は硝酸酸性,電気加熱原子吸光法及びICP質量分析法を適用する場合は,
硝酸酸性とし,フレーム原子吸光法及び電気加熱原子吸光法の場合には,0.1〜1 mol/L,ICP発光分光分析
法においては,0.1〜0.5 mol/Lとする。また,ICP質量分析法の場合には,硝酸酸性,濃度は0.1〜0.5 mol/L
とする。ただし,いずれの場合も検量線作成時の場合とほぼ同じ濃度とする。
6.6.1
留意事項
ICP発光分光分析法の場合,硫酸酸性では試料導入量が少なく感度が悪くなることがあるので,6.5の適
用は,やむを得ない場合だけとする。
7
pH
7.1
一般事項
pHの測定には,JIS Z 8802によるガラス電極法又はpHプロセス用分析装置による測定方法を適用する。
pHは試料採取後,直ちに測定する。
7.2
ガラス電極法
ガラス電極を用いたpH計によってpHを測定する。
7.2.1
試薬
試薬は,次による。
a) 水 JIS K 0557で規定するA2又はA3の水。ほう酸塩pH標準液及び炭酸塩pH標準液を調製する場
合は,4.7 a) の二酸化炭素を含まない水を用いる。
b) 二しゅう酸三水素カリウム二水和物 JIS K 8474で規定するもの。
c) フタル酸水素カリウム JIS K 8809で規定するフタル酸水素カリウムのpH標準液用。
d) りん酸二水素カリウム JIS K 9007で規定するりん酸二水素カリウムのpH標準液用。
e) りん酸水素二ナトリウム JIS K 9020で規定するりん酸水素二ナトリウムのpH標準液用。
f)
四ほう酸ナトリウム十水和物 JIS K 8866で規定する四ほう酸ナトリウム十水和物のpH標準液用。
g) 炭酸水素ナトリウム JIS K 8622で規定する炭酸水素ナトリウムのpH標準液用。
h) 炭酸ナトリウム JIS K 8625で規定する炭酸ナトリウムのpH標準液用。
i)
pH標準液 pH標準液は,次による。
1) 調製pH標準液 調製pH標準液は,JIS Z 8802の7.3.2(調製方法)による。
2) 認証pH標準液 国家計量標準とのトレーサビリティが確認された認証pH標準液は,第2種を用
いる。
3) pH標準液の各温度でのpHは,表1及び表2による。この表に記載しない温度におけるpHは,補
間して求める。
4) 各pH標準液は,長期間保存するとpHが変化することがあるので長期間保存したものは使用しない。
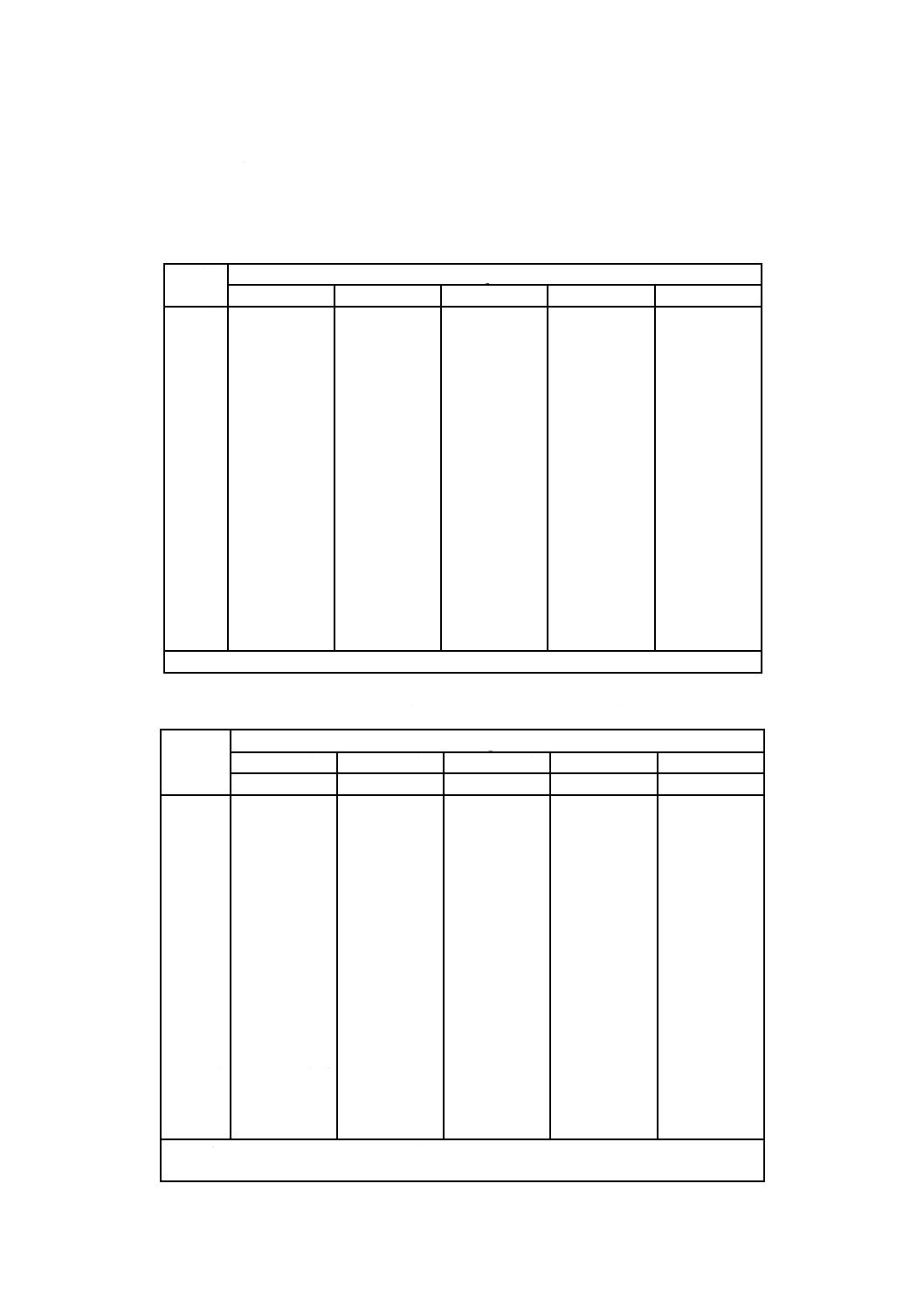
11
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
特に,ほう酸塩pH標準液及び炭酸塩pH標準液は,容易に大気中の二酸化炭素を吸収し,pHが低
下するので注意する。
5) 各pH標準液は,一度使用したもの及び大気中に開放して放置したものは使用しない。
表1−調製pH標準液の各温度におけるpHの典型値
温度
℃
pH
しゅう酸塩
フタル酸塩
中性りん酸塩
ほう酸塩
炭酸塩a)
0
5
10
15
20
25
30
35
38
40
45
50
55
60
70
80
90
95
1.67
1.67
1.67
1.67
1.68
1.68
1.69
1.69
−
1.70
1.70
1.71
1.72
1.73
1.74
1.77
1.80
1.81
4.01
4.01
4.00
4.00
4.00
4.01
4.01
4.02
−
4.03
4.04
4.06
4.08
4.10
4.12
4.16
4.20
4.23
6.98
6.95
6.92
6.90
6.88
6.86
6.85
6.84
−
6.84
6.83
6.83
6.84
6.84
6.85
6.86
6.88
6.89
9.46
9.39
9.33
9.27
9.22
9.18
9.14
9.10
−
9.07
9.04
9.01
8.99
8.96
8.93
8.89
8.85
8.83
10.32
(10.25)
10.18
(10.12)
(10.07)
10.02
(9.97)
(9.93)
9.91
−
−
−
−
−
−
−
−
−
注a) 括弧内の値は,2次補間値を示す。
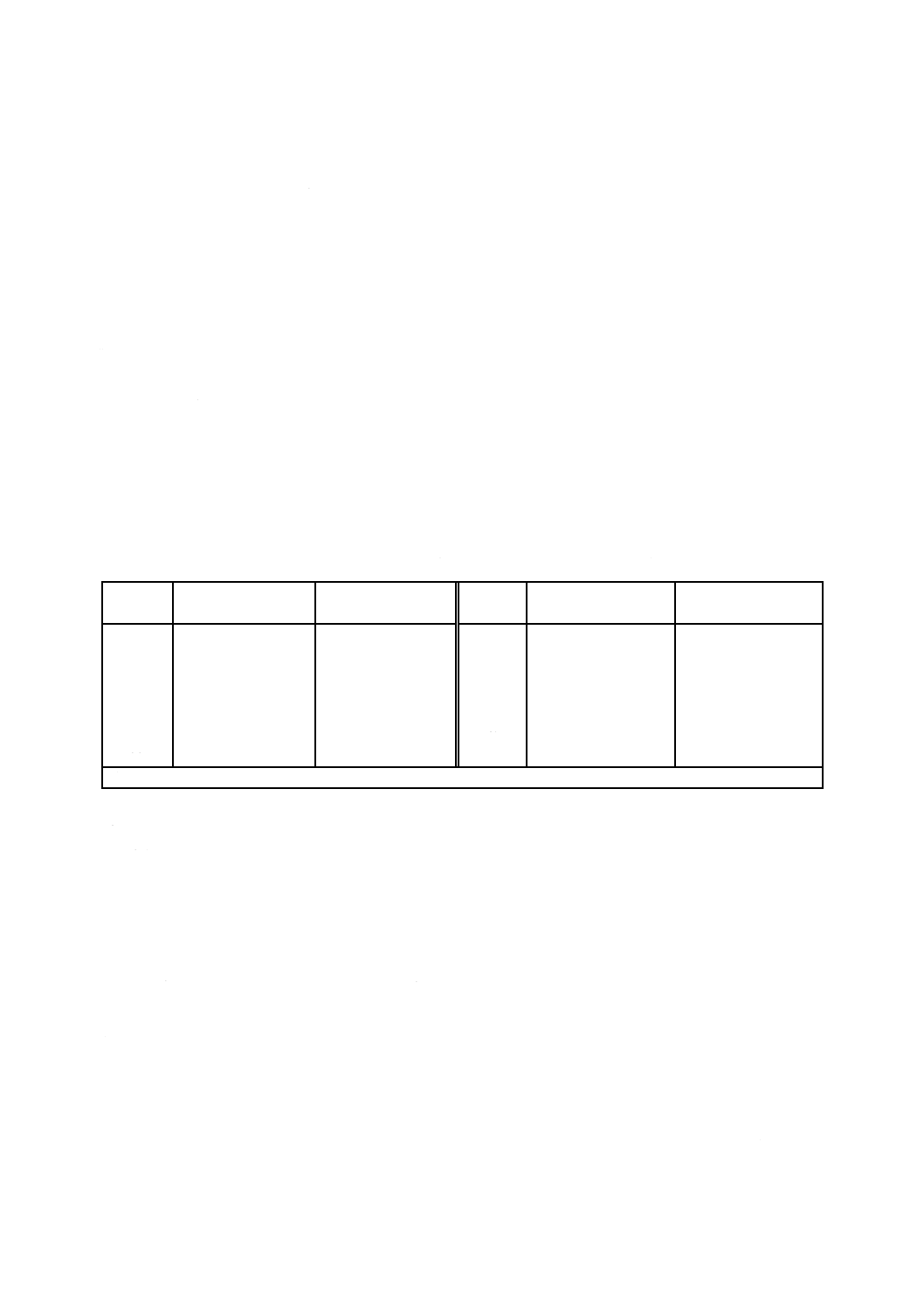
表2−認証pH標準液の各温度におけるpHの典型値a)(参考)
温度
℃
pH
しゅう酸塩
フタル酸塩
中性りん酸塩
ほう酸塩
炭酸塩
第2種
第2種
第2種
第2種
第2種
0
5
10
15
20
25
30
35
38
40
45
50
55
60
70
80
90
95
1.67
1.67
1.67
1.67
1.68
1.68
1.68
1.69
1.69
1.69
1.70
1.71
1.72
1.72
1.74
1.77
1.79
1.81
4.00
4.00
4.00
4.00
4.00
4.01
4.02
4.02
4.03
4.04
4.05
4.06
4.08
4.09
4.13
4.16
4.20
4.23
6.98
6.95
6.92
6.90
6.88
6.86
6.85
6.84
6.84
6.84
6.83
6.83
6.83
6.84
6.84
6.86
6.88
6.89
9.46
9.40
9.33
9.28
9.22
9.18
9.14
9.10
9.08
9.07
9.04
9.01
8.98
8.96
8.92
8.88
8.85
8.83
10.32
10.24
10.18
10.12
10.06
10.01
9.97
9.92
−
9.89
9.86
9.83
−
−
−
−
−
−
注a) OIML recommendation (R054-e81) に記載の値を小数点以下2桁に丸めたものである。JIS
Z 8802による。
12
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7.2.2
器具及び装置
器具及び装置は,次による。
a) pH計 繰返し精度が±0.05の精度のもの(JIS Z 8802で規定する形式II相当)を用いる。
b) 温度計 許容差±1 ℃のもの。JIS B 7411規格群で規定する一般用ガラス製棒状温度計の50度温度計
又はJIS C 1602で規定する熱電対を用いる。
7.2.3
pH計の校正
pH計の校正は,次による。
a) pH計の電源を入れ,検出部(ガラス電極,参照電極,温度補償電極など)を取り付ける。電極の特性
が安定するよう校正作業に入る前に電極が30分間以上,水に浸された状態であることを確認する。
b) 電極を水で洗浄し,中性りん酸塩pH標準液を入れたビーカに浸す。温度補償用ダイヤル又はデジタ
ルスイッチの設定のあるものは目盛値を,中性りん酸塩pH標準液の温度に合わせる。
c) 中性りん酸塩pH標準液の温度に対応するpH(表1又は表2)に調整ダイヤルを調節して合わせる。
d) 電極を水で洗浄し,試料のpHが7以下の場合は,フタル酸塩pH標準液又はしゅう酸塩pH標準液を
入れたビーカに浸す。スパン調整ダイヤルを調節して使用したpH標準液の温度に対応するpH(表1
又は表2)に合わせる。試料のpHが7を超える場合は,ほう酸塩pH標準液又は炭酸塩pH標準液を
用い,同じ操作でpH標準液の温度に対応するpHに合わせる。
e) 再びb)〜d) の操作を行い,pHの指示値がpH標準液の温度に対応するpHに±0.05であることを確認
し,逸脱する場合は,この操作を繰り返す。
7.2.4
操作
操作は,次による。
a) 校正したpH計の検出部を水で繰り返し3回以上洗い,きれいな柔らかい紙などで拭っておく。
b) 試料をビーカにとり液温を25±2 ℃に調節し,これに検出部を浸す。温度補償用ダイヤル又はデジタ
ルスイッチの設定のあるものは目盛値を25 ℃に合わせた後,pHを測定する。
c) 検出部を取り出し,水で繰り返し3回以上洗い,きれいな柔らかい紙などで拭っておく。
d) 再び試料をビーカにとり液温を25±2 ℃に調節し,これに検出部を浸し,pHを測定する。
e) 再びc) 及びd) の操作を行って3回の測定値が±0.1で一致した測定値を平均して,試料のpHを算出
する。
7.2.5
留意事項
a) 長く乾燥状態にあったガラス電極は,あらかじめ水に浸して平衡に達してから使用する。
b) ガラス電極が汚れている場合は,必要に応じて洗剤及び塩酸(1+20)(JIS K 8180で規定する塩酸を
用いて調製する。)などで短時間洗い,更に流水で十分に洗う。電極の取扱いは製造業者の取扱説明書
による。
c) 参照電極の汚れの除去はガラス電極と同じ操作で行い,内部液(塩化カリウム溶液)の交換などは取
扱説明書を参照する。液絡部の汚染を避けるため,電解液には2 cm以上の水位差に相当する静水圧が
必要である。
d) pH校正で電極類をすすぎ洗いするときは,ガラス電極及び参照電極,温度補償電極などが同時に入る
大きさのビーカに純水を入れ,その中で行う。
e) 校正において,pHの指示値がpH標準液の温度に対応するpHに対し,形式Iでは±0.02,形式IIIで
は±0.1であることを確認し,逸脱する場合は,この操作を繰り返す。
f)
JIS B 8223で規定するpHについては,25 ℃におけるpHを示してあるので,25 ℃の起電力が測定で
13
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
きるようにpH計の温度補償回路を調節する。
g) アンモニア,りん酸ナトリウム,ヒドラジンなどpH-濃度-温度曲線に沿った試料水の温度補償回路の
設定で実用上問題のない補償ができる場合には,pH計の起電力の温度依存性及び試料のpHの温度依
存性の両方に対応した二重の温度補償回路をもったpH計が使用できる。
h) pHは試料の温度によって異なるので,試料の温度変動は±2 ℃にする。
i)
緩衝性が乏しい試料は,容易にpHが変化するため,±0.1の繰返し精度が得られない場合がある。こ
の場合は,pHが±0.2で一致する値を平均してpHを算出する。また,大気中の二酸化炭素で容易に
pHが変動する場合には,流液形の測定セルを使用するとよい。
j)
試料のpHが11以上の場合には,通常のガラス電極ではアルカリ誤差を生じ,測定値が低くなる。特
にアルカリ金属イオンの濃度が高い場合は誤差が大きくなるので,アルカリ誤差が少ない電極を用い,
炭酸塩を含まない0.1 mol/L水酸化ナトリウム溶液又は25 ℃の飽和水酸化カルシウム溶液をpH標準
液として用いてpH計の校正を行い,試料のpHを測定する。
0.1 mol/L水酸化ナトリウム溶液又は飽和水酸化カルシウム溶液は,大気中の二酸化炭素を吸収して
容易にpHが低下するので使用の都度,調製する。
表3に0.1 mol/L水酸化ナトリウム溶液及び飽和水酸化カルシウム溶液の各温度におけるpHを示す。
表3−0.1 mol/L水酸化ナトリウム溶液及び飽和水酸化カルシウム溶液の各温度におけるpH
温度
℃
0.1 mol/L水酸化
ナトリウム溶液
飽和水酸化
カルシウム溶液a)
温度
℃
0.1 mol/L水酸化
ナトリウム溶液
飽和水酸化
カルシウム溶液
0
5
10
15
20
25
30
13.8
13.6
13.4
13.2
13.1
12.9
12.7
13.43
13.21
13.00
12.81
12.63
12.45
12.30
35
40
45
50
55
60
12.6
12.4
12.3
12.2
12.0
11.9
12.14
11.99
11.84
11.70
11.58
11.45
注a) 25 ℃における飽和水酸化カルシウム溶液
7.3
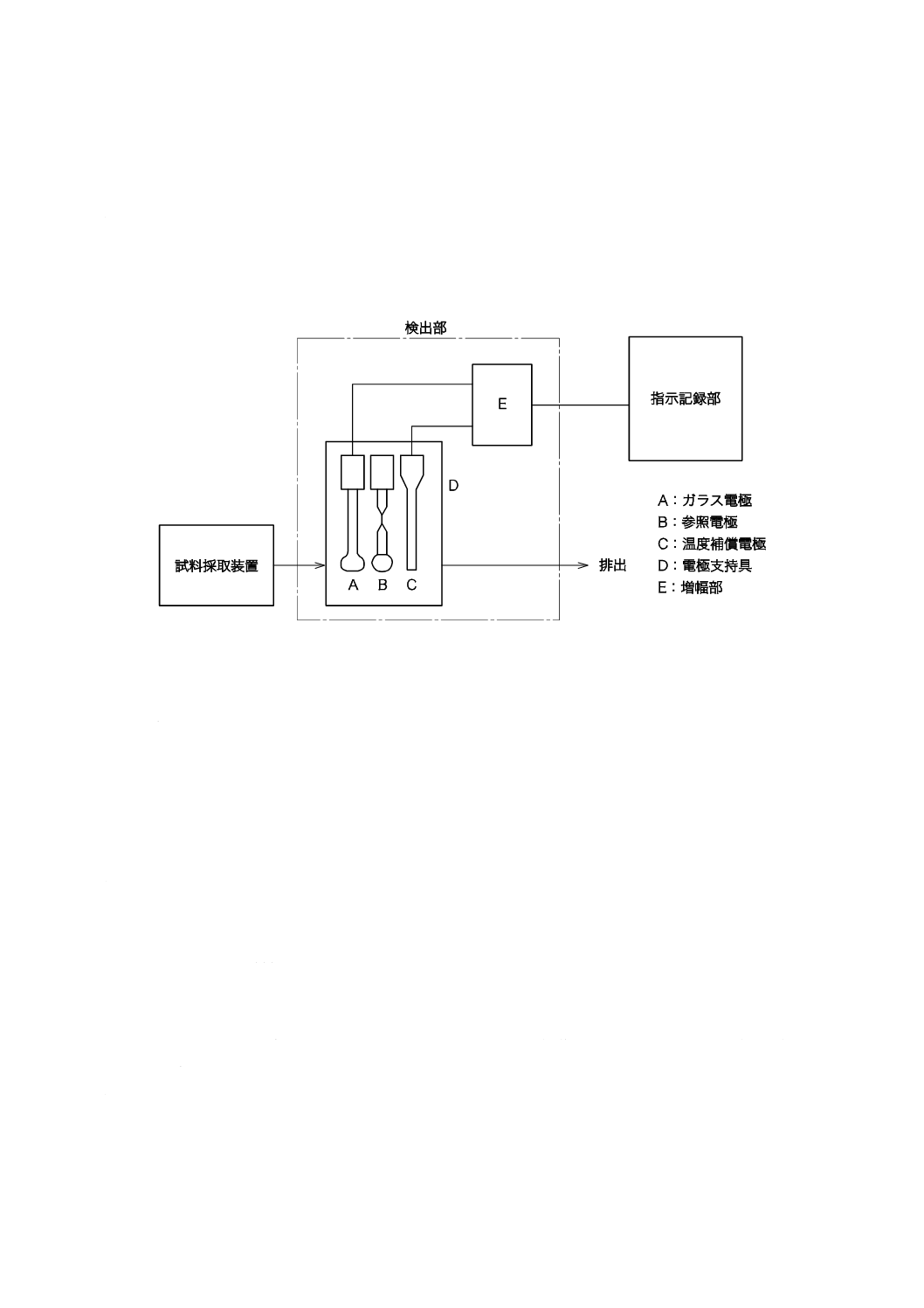
pHプロセス用分析装置による測定方法
測定範囲:pH 0〜14,繰返し精度:±0.1 pH
7.3.1
測定原理
ガラス電極を用いたpHプロセス用分析装置によってpHを連続的に測定する。
7.3.2
器具及び装置
器具及び装置は,次による。
a) pHプロセス用分析装置 箇条3 a) で規定するプロセス用分析装置で,JIS K 0802で規定するpH自
動計測器の性能及び構造を備えたもの。その構成例を図3に示す。
b) 熱交換器又は恒温槽 試料を25±2 ℃に調節できるもの。
7.3.3
校正
pHプロセス用分析装置の校正は,7.2.3による。
7.3.4
操作
校正が終了したpHプロセス用分析装置の検出部を試料で十分に洗浄した後,25±2 ℃に調節した試料
を所定の流量で導入し,連続測定を行う。
14
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7.3.5
点検・整備
点検・整備は,定期的に次のことを行うこととし,手順の詳細は製造業者の提供する取扱説明書による。
a) 試料流量が所定量であることを確認する。
b) 電極の汚れがないことを確認する。
c) 参照電極の内部液を補充又は交換する。
d) 温度補償電極を点検する。
図3−pHプロセス用分析装置の構成の一例
8
電気伝導率
8.1
一般事項
電気伝導率は,溶液がもつ電気抵抗率(Ω・m)の逆数に相当し,S/mの単位で表す。また,電気伝導度
は,溶液のもつ電気抵抗(Ω)の逆数に相当し,Sの単位で表す。
水の試験では,温度25 ℃の値を用い,mS/m又はμS/cmで示す。1 μS/cmは,0.1 mS/mに相当する。試
料の電気伝導率が1 mS/m(25 ℃)以下の測定の場合には,JIS K 0552を適用する。
電気伝導率の試験は,一般試験,酸電気伝導率試験及び電気伝導率プロセス用分析装置による測定に区
分する。
8.2
一般試験
電気伝導率計を用いて,試料の電気伝導率を測定する。
測定範囲:0.005〜1 000 mS/m(25 ℃)
8.2.1
試薬
試薬は,次による。
a) 水 JIS K 0557で規定するA2又はA3の水。ただし,電気伝導率0.2 mS/m[2 µS/cm](25 ℃)以下
のものを20±2 ℃に調節して用いる。
b) 塩化カリウム JIS K 8121で規定する塩化カリウム(電気伝導率測定用)をめのう乳鉢で粉末にし,
500 ℃で約4時間加熱してデシケータ中で放冷する。
c) 塩化カリウム標準液(C) b) の塩化カリウム0.744 gをはかりとり,少量のa) の水に溶かし,全量
15
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
フラスコ1 000 mLに移し入れ,a) の水を標線まで加える。
d) 塩化カリウム標準液(E) c) の塩化カリウム標準液(C)10 mLを全量フラスコ1 000 mLにとり,
a) の水を標線まで加える。
これらの塩化カリウム標準液は,共栓ポリエチレン瓶又は共栓ほうけい酸ガラス瓶に密栓して保存
する。
8.2.2
器具及び装置
器具及び装置は,次による。
a) 電気伝導率計 目的とする電気伝導率に応じて,表4に示すセル定数のものを用意する。
b) 温度計 JIS C 1604のクラスB又は,JIS C 1611の1.0級に相当する温度検出素子を用いたもので,
試料の温度を±1 ℃で測定できるもの。
8.2.3
操作
操作は,次による。
a) あらかじめ電気伝導率計の電源を入れておく。試料の電気伝導率に応じて表4に示すセル定数をもっ
た電極を用い,水でセルを2〜3回洗う[特に汚れている場合には,塩酸(1+100)に浸し,更に流水
で十分に洗い,最後に水で2〜3回洗う]。
b) このセルを試料で2〜3回洗った後,試料を満たし,25±2 ℃に保って電気伝導率の測定を行う。測定
値が±3 %で一致するまで試料を数回取り替えて測定を繰り返し,その電気伝導率を求める。
表4−セル定数と測定範囲
セル定数a)
測定範囲
m−1
cm−1
mS/m
µS/cm
1
10
100
1 000
2 000
0.01
0.1
1
10
20
0.005 〜
20
0.005 〜
1 000
0.1
〜 10 000
1
〜 100 000
10
〜 100 000
0.05 〜
200
0.05 〜
10 000
1
〜
100 000
10
〜 1 000 000
100
〜 1 000 000
注a) セル定数m−1×0.01=cm−1
8.2.4
留意事項
a) 精度を特に必要としない場合には,温度補償回路を組み入れた電気伝導率計を用いるか,又は温度換
算式を用いてもよい。電気伝導率は,温度によって変化し,1 ℃の上昇で約2 %大きくなる。ただし,
電気伝導率が0.1 mS/m(1 µS/cm)以下になると,水の解離によって生じる水素イオン及び水酸化物
イオンの影響が大きくなるので,この換算式は適用できない。
b) 試料の電気伝導率が1 mS/m(25 ℃)未満の場合には,±3 %で一致しないことがあるので,JIS K 0552
に従って試験するか,又は流液形のセルを用いる。
c) セル定数の確認方法 セル定数を確認しようとする場合には,セルを水で2〜3回洗い,次に,塩化カ
リウム標準液(セル定数に応じ,表5の測定範囲の塩化カリウム標準液を用いる。)で,2〜3回洗っ
た後,その塩化カリウム標準液を満たす。このセルを25±0.5 ℃に保ち,電気伝導度を測定する。同
じ塩化カリウム標準液を数回入れ替えて測定を行い,測定値が±3 %で一致するまで繰り返す。
測定された値から次の式によってセル定数を算出する。

16
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
XO
O
H
KCl
2
L
L
L
J
+
=
ここに,
J: セル定数(m−1)
LXO: 測定した電気伝導度(mS)。ただし,電気伝導度の指示
がµSになっているときは,µS×000
1
1の値を用いる。
LKCl: 使用した塩化カリウム標準液のこの温度における電気
伝導率(mS/m)
LH2O: 塩化カリウム標準液の調製に用いた水のこの温度にお
ける電気伝導率(mS/m)
表5−塩化カリウム標準液(C及びE)の電気伝導率
塩化カリウム標準液
℃
mS/m
µS/cm
C
0
18
25
77.4
122.0
140.9
774
1 220
1 409
E
25
1.49
14.9
8.3
酸電気伝導率
揮発性物質処理を行っているボイラにおいて,発生蒸気,蒸気凝縮水及び給水中の揮発性物質以外の塩
類の存在を知るために,これらを水素イオン形に変換した強酸性陽イオン交換樹脂を充塡したカラムに通
して除去した後の電気伝導率を測定する。
揮発性物質,例えば,ヒドラジニウムイオン(ヒドラジン),モルホリン(テトラヒドロキシ-1,4-オキサ
ジン),アンモニウムイオンなどは水素イオン形に変換した強酸性陽イオン交換樹脂によってイオン交換さ
れ,これらによる電気伝導率の増加分が消去される。しかし,これらの揮発性物質以外の陽イオンは,水
素イオン形の強酸性陽イオン交換樹脂の水素イオンとイオン交換反応を行い,陽イオンと当量の水素イオ
ンを生成する。この水素イオンによる電気伝導率を測定して微量に存在する塩類の濃度を推定する。試験
は,次による。
測定範囲:0.005〜1 mS/m(25 ℃)
8.3.1
試薬
試薬は,次による。
a) 塩酸(1+11) JIS K 8180で規定する塩酸を用いて調製する。
b) 強酸性陽イオン交換樹脂 次のように水素イオン形に変換したものを用いる。
強酸性陽イオン交換樹脂の適量を水(イオン交換樹脂量の約3倍量)に浸し,12時間以上放置した
後,適切なカラム(イオン交換樹脂を充塡したとき,イオン交換樹脂層の高さが約600 mmになるよ
うなカラム)に水で移し入れる。強酸性陽イオン交換樹脂1 Lにつきa) の塩酸(1+11)20 Lを,8
〜10 L/h・(L-樹脂) で流し,引き続き水3 Lを同じ流量で流して水素イオン形に変換する。次に,水を
約20 L/h・(L-樹脂) の割合で80 L/(L-樹脂) 以上流して洗浄する。
これを図4のイオン交換樹脂カラムに気泡が入らないように水で充塡する。
8.3.2
器具及び装置
器具及び装置は,次による。
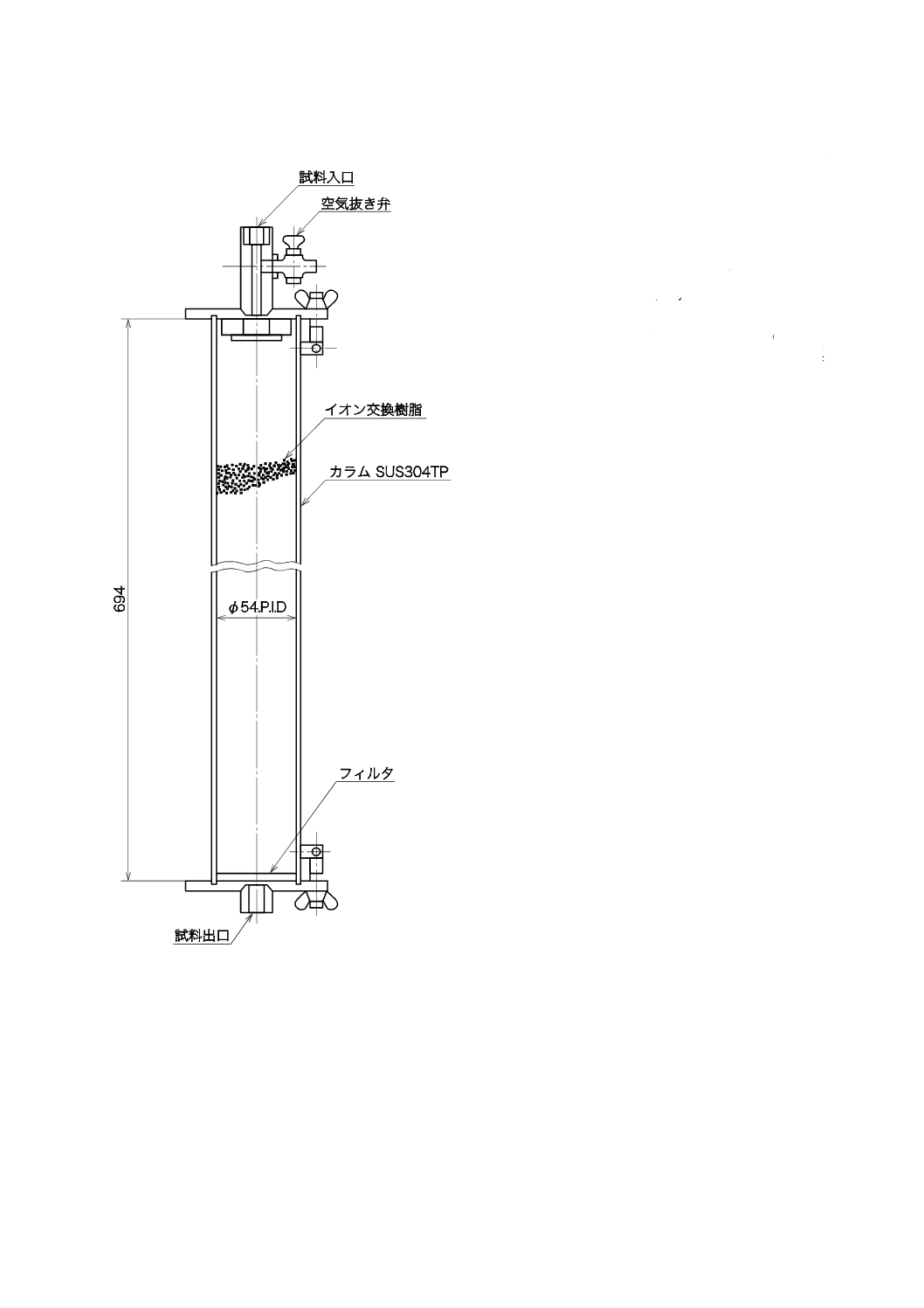
a) イオン交換樹脂カラム イオン交換樹脂カラムの一例を図4に示す。イオン交換樹脂を充塡したとき,
17
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
イオン交換樹脂層の高さが約600 mmになるようなカラムを選ぶ。
b) 電気伝導率計 電気伝導率0.005〜1 mS/m(0.05〜10 µS/cm)(25 ℃)を測定できるもので,セル定数
0.8〜12 m−1(0.008〜0.12 cm−1)の流液形検出器が使用できるもの。
8.3.3
操作
操作は,次による。
a) 水素イオン形に変換した強酸性陽イオン交換樹脂を充塡したイオン交換樹脂カラムに試料を12〜15
L/h・(L-樹脂) の割合で連続的に流す。
b) a) の流出液を流液形検出器に導入し,指示値が安定した後,電気伝導率を測定する。
8.3.4
留意事項
a) 強酸性陽イオン交換樹脂の代わりに,カチオン交換膜を用いた電気式カチオン交換器を使用してもよ
い。
b) 強酸性陽イオン交換樹脂を水素イオン形に変換する際に,水洗浄の洗液が,9.2.1 a) のメチルレッド-
ブロモクレゾールグリーン混合溶液で青い色になることで洗浄の終了を判断してもよい。
c) 強酸性陽イオン交換樹脂は粒子径355〜1 180 μmのものでジビニルベンゼンの含量約8 %のものを用
いている。
18
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
単位 mm
常用圧力:0.1 MPa以下
容量:1.6 L
イオン交換樹脂の充塡量:1.5 L
流量:12〜15 L/h・(L-樹脂)
材料:ステンレス鋼(SUS304)製
図4−イオン交換樹脂カラムの一例
8.4
電気伝導率プロセス用分析装置による測定方法
測定範囲:0.005〜0.2 mS/m,0.005〜10 mS/m,0.005〜100 mS/m,繰返し精度:最大目盛値の±1.5 %以
下。
8.4.1
測定原理
電気伝導率プロセス用分析装置によって給水,ボイラ水などの電気伝導率又は酸電気伝導率を連続的に
測定する。

19
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8.4.2
器具及び装置
器具及び装置は,次による。
a) 電気伝導率プロセス用分析装置 箇条3 a) で規定するプロセス用分析装置で,検出部,指示部及び記
録部で構成する。その構成の一例を図5に示す。電気伝導率が0.1 mS/m以下の場合には,JIS K 0552
で規定する二重温度補償ができる方式のものを用いる。
図5−電気伝導率プロセス用分析装置の構成の一例
8.4.3
校正
電気伝導率プロセス用分析装置が安定状態に達した後,次による。
a) スパン校正 校正用電気抵抗で指示値を所定の値に調節する。
8.4.4
操作
試料採取装置から直接流出した流出液,又は水素イオン形に変換した強酸性陽イオン交換樹脂を充塡し
たイオン交換樹脂カラムを通過した流出液を流液形検出器に連続的に導入し,電気伝導率を測定する。
8.4.5
点検・整備
点検・整備は,定期的に次のことを行うこととし,手順の詳細は製造業者の提供する取扱説明書による。
a) 試料の流量が所定量であることを確認する。
b) セル(検出部)の汚れがないことを確認する。
c) 電気伝導率プロセス用分析装置で連続的に酸電気伝導率を測定する場合,強酸性陽イオン交換樹脂を
定期的に交換・再生する。
8.4.6
留意事項
a) 電気伝導率プロセス用分析装置で連続的に酸電気伝導率を測定する場合,強酸性陽イオン交換樹脂の
交換・再生頻度については,次の計算方法で推定できる。
計算の条件(一例)
強酸性陽イオン交換樹脂層に流入する試料中のアンモニウムイオンの濃度NH4+:1.4 mg/L
強酸性陽イオン交換樹脂の交換容量:1.9 mol/L-樹脂{1.9 g当量/L-樹脂}
強酸性陽イオン交換樹脂層の流量:15 L/h・(L-樹脂)
強酸性陽イオン交換樹脂の充塡量:1.5 L
計算(一例)
通水流量(L/日)
540
24
5.1
15
=
×
×
=
予想通水量(L)
3
3
10
65
.
25
4.1
7.0
10
18
9.1
5.1
×
=
×
×
×
×
=
ここに,
18: アンモニウムイオンの分子量(g/mol)
0.7: イオン交換樹脂の使用率 70 %
図4の樹脂カラムを使用した場合
通水可能日数(日)
5.
47
540
10
65
.
25
3=
×
=
20
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
したがって,約47日ごとに再生を繰り返すことが必要になる。
9
酸消費量
9.1
一般事項
酸消費量は,水に溶けている炭酸水素塩,炭酸塩,水酸化物などによるアルカリを所定のpHに中和す
るのに要する水素イオンの量(酸の量)を,試料1 Lについてのmmol数で表すか,又は水素イオン(酸)
に相当する炭酸カルシウムの量に換算して,試料1 Lについてのmg数で表す。
酸消費量を酸消費量(pH 4.8)と酸消費量(pH 8.3)とに区分する。
9.2
酸消費量(pH 4.8)
酸消費量(pH 4.8)は,試料に指示薬としてメチルレッド-ブロモクレゾールグリーン混合溶液を加え,
10 mmol/L硫酸で滴定して求める。
9.2.1
試薬
試薬は,次による。
a) メチルレッド-ブロモクレゾールグリーン混合溶液 JIS K 8896で規定するメチルレッド0.02 gとJIS
K 8840で規定するブロモクレゾールグリーン0.1 gとをJIS K 8102で規定するエタノール(95)100 mL
に溶かす。
b) 50 mmol/L硫酸 JIS K 8951で規定する硫酸3 mLを,あらかじめ水100 mLを入れたビーカに加えて
よくかき混ぜ,放冷後,水を加えて1 Lとする。この溶液は,次によって標定して用いる。
1) JIS K 8005で規定する容量分析用標準物質の炭酸ナトリウムを600 ℃で約1時間加熱した後,デシ
ケータ中で放冷する。その1.06 gを1 mgの桁まではかりとり,水に溶かして全量フラスコ200 mL
に移し入れ,水を標線まで加える。
2) この20 mLをビーカにとり,指示薬としてメチルレッド-ブロモクレゾールグリーン混合溶液3〜5
滴を加えた後,この50 mmol/L硫酸で滴定する。溶液の色が灰紫になったら,煮沸して二酸化炭素
を追い出し,放冷後,溶液の色が灰紫になるまで滴定を続ける。
3) 滴定に要した50 mmol/L硫酸のmL数から,次の式によって50 mmol/L硫酸のファクタ( f )を算
出する。
30
005
.0
1
200
20
100
×
×
×
×
=
x
b
a
f
ここに,
a: 炭酸ナトリウムの量(g)
b: 炭酸ナトリウムの純度(%)
x: 滴定に要した50 mmol/L硫酸(mL)
0.005 30: 50 mmol/L硫酸1 mLの炭酸ナトリウム相当量(g)
c) 10 mmol/L硫酸 50 mmol/L硫酸200 mLを全量フラスコ1 000 mLにとり,水を標線まで加える。フ
ァクタは50 mmol/L硫酸の値を用いる。
9.2.2
操作
操作は,次による。
a) 試料100 mLをビーカにとり,指示薬としてメチルレッド-ブロモクレゾールグリーン混合溶液3〜5
滴を加える。ただし,試料に濁りがあるときはろ紙5種Bでろ過するか又は遠心分離してその上澄み
液を用いる。また,残留塩素などの酸化性物質が共存する場合には,チオ硫酸ナトリウム溶液(0.1
mol/L)で還元してから加える。
21
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) この溶液を緩やかにかき混ぜながら10 mmol/L硫酸で,溶液の色が青から灰紫(pH 4.8)に変わるま
で滴定する。
c) 着色した試料など,指示薬による変色が明らかでない場合には,指示薬に代えてpH計を使用し,pH
と10 mmol/L硫酸の滴定量とによる滴定曲線を作成して,pH 4.8における10 mmol/L硫酸のmL数を
求める。
d) 次の式によって酸消費量(pH 4.8)を算出する。
mmol/Lで表す場合
02
.0
000
1
×
×
×
=
V
f
a
A
ここに,
A: 酸消費量(pH 4.8)(mmol/L)
a: 滴定に要した10 mmol/L硫酸(mL)
f: 10 mmol/L硫酸のファクタ
V: 試料(mL)
0.02: 10 mmol/L硫酸1 mLの水素イオン相当量(mmol)
CaCO3:mg/Lで表す場合
001
.1
000
1
×
×
×
=
V
f
a
B
ここに,
B: 酸消費量(pH 4.8)(CaCO3:mg/L)
a: 滴定に要した10 mmol/L硫酸(mL)
f: 10 mmol/L硫酸のファクタ
V: 試料(mL)
1.001: 10 mmol/L硫酸1 mLの炭酸カルシウム相当量(mg)
9.3
酸消費量(pH 8.3)
酸消費量(pH 8.3)は,試料に指示薬としてフェノールフタレイン溶液を加え,10 mmol/L硫酸で滴定し
て求める。
9.3.1
試薬
試薬は,次による。
a) フェノールフタレイン溶液(5 g/L) JIS K 8799で規定するフェノールフタレイン0.5 gをとり,JIS
K 8102で規定するエタノール(95)50 mLに溶かし,水を加えて100 mLとし,この溶液の色が僅か
に赤に呈色するまで水酸化ナトリウム溶液(20 mmol/L)を滴加する。
b) 10 mmol/L硫酸 9.2.1 c) による。
9.3.2
操作
操作は,次による。
a) 試料100 mLをビーカにとり,指示薬としてフェノールフタレイン溶液(5 g/L)3〜5滴を加える。
b) この溶液を緩やかにかき混ぜながら10 mmol/L硫酸で,溶液の赤が消えるまで滴定する。
c) 指示薬による変色が明らかでない場合は,指示薬に代えてpH計を使用し,pHと10 mmol/L硫酸の滴
定量とによる滴定曲線を作成して,pH 8.3における10 mmol/L硫酸のmL数を求める。
d) 次の式によって酸消費量(pH 8.3)を算出する。
mmol/Lで表す場合
02
.0
000
1
×
×
×
=
V
f
a
A
ここに,
A: 酸消費量(pH 8.3)(mmol/L)
22
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a: 滴定に要した10 mmol/L硫酸(mL)
f: 10 mmol/L硫酸のファクタ
V: 試料(mL)
0.02: 10 mmol/L硫酸1 mLの水素イオン相当量(mmol)
CaCO3:mg/Lで表す場合
001
.1
000
1
×
×
×
=
V
f
a
B
ここに,
B: 酸消費量(pH 8.3)(CaCO3:mg/L)
a: 滴定に要した10 mmol/L硫酸(mL)
f: 10 mmol/L硫酸のファクタ
V: 試料(mL)
1.001: 10 mmol/L硫酸1 mLの炭酸カルシウム相当量(mg)
10
硬度
10.1
一般事項
硬度は,水中のカルシウムイオン及びマグネシウムイオンの量を,これに対応する炭酸カルシウムの量
に換算して試料1 Lについてのmg数で表す。
硬度は,全硬度,カルシウム硬度及びマグネシウム硬度に区分する。カルウムイオン及びマグネシウム
イオンの濃度をキレート滴定法,フレーム原子吸光法,ICP発光分光分析法,ICP質量分析法で求め,そ
れらの測定値から全硬度,カルシウム硬度及びマグネシウム硬度を求める。
10.2
全硬度
10.2.1
キレート滴定法
10.2.1.1 試薬
試薬は,次による。
a) シアン化カリウム溶液(100 g/L) JIS K 8443で規定するシアン化カリウム10 gを水に溶かして100
mLとする。ポリエチレン瓶に保存する。
警告 シアン化カリウムは有毒である。取扱い及び廃液の処理は,法令に従い,安全及び健康に対
する適切な措置を取らなければならない。シアン化カリウムを含む溶液は,有毒なシアン化
水素ガスが発生するため,酸性にしてはならない。
b) 塩化ヒドロキシルアンモニウム溶液(100 g/L) JIS K 8201で規定する塩化ヒドロキシルアンモニウ
ム10 gを水に溶かして100 mLとする。
c) 塩化アンモニウム-アンモニア緩衝液(pH 10) JIS K 8116で規定する塩化アンモニウム67.5 gにJIS
K 8085で規定するアンモニア水570 mLを加え,水で1 Lとする。この溶液は密栓して冷所に保存す
る。
d) エリオクロムブラックT溶液(5 g/L) JIS K 8736で規定するエリオクロムブラックT[1-(1-ヒド
ロキシ-2-ナフチルアゾ)-6-ニトロ-2-ナフトール-4-スルホン酸ナトリウム]0.5 gをJIS K 8891で規定
するメタノール100 mLに溶かし,JIS K 8201で規定する塩化ヒドロキシルアンモニウム0.5 gを加え
る。着色瓶に入れ,密栓して保存する。
e) 10 mmol/L EDTA溶液 JIS K 8107で規定するエチレンジアミン四酢酸二水素二ナトリウム二水和物
を80 ℃で約5時間加熱し,デシケータ中で放冷する。その3.772 gをとり,少量の水に溶かし,全量
フラスコ1 000 mLに移し入れ,水を標線まで加える。
10.2.1.2 操作
23
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
操作は,次による。
a) 懸濁物質が含まれる試料などでは,ろ過又は遠心分離によって懸濁物を除去した後,適量(マグネシ
ウムとカルシウムとの合量をカルシウムに換算して5 mg以下とする。)をビーカにとり,水を加えて
約50 mLとする。
b) シアン化カリウム溶液(100 g/L)0.5 mL,塩化ヒドロキシルアンモニウム溶液(100 g/L)数滴及び塩
化アンモニウム-アンモニア緩衝液(pH 10)1 mLを加える。
c) 指示薬としてエリオクロムブラックT溶液(5 g/L)2,3滴を加える。
d) 10 mmol/L EDTA溶液で,溶液の赤みが消えて青になるまで滴定する。エリオクロムブラックTの変
色は遅いので,変色点近くではよくかき混ぜながらゆっくり滴定する。
e) 次の式によって全硬度を算出する。
001
.1
000
1
×
×
=
V
b
H
ここに,
H: 全硬度(CaCO3:mg/L)
b: 滴定に要した10 mmol/L EDTA溶液(mL)
V: 試料(mL)
1.001: 10 mmol/L EDTA溶液1 mLの炭酸カルシウム相当量
(mg)
10.2.1.3 留意事項
a) EDTAによる滴定は,pHが低すぎるとカルシウム,マグネシウムはいずれも定量的に反応しない。ま
た,pHが高すぎるとマグネシウムは水酸化物となり,EDTAと反応しなくなる。そのため,滴定時に
は緩衝液を加えてpHを約10に調節する。
b) 滴定に際しての,カルシウム,マグネシウム,EDTA及びEBT(エリオクロムブラックT)の錯体生
成の強さは,次のようになる。
Ca-EDTA > Mg-EDTA > Mg-EBT(赤)> Ca-EBT
したがって,滴定の開始時は,指示薬として添加したごく少量のEBTは,Mg-EBT(赤)となる。
EDTAの滴加に従い,まず,遊離のカルシウム,次いでマグネシウムがEDTAと結合し,当量点では,
次の反応で青に変色する。
Mg-EBT(赤)+ EDTA → Mg-EDTA + EBT(青)
ただし,試料中にマグネシウムが存在しなければ,終点での変色はCa-EBTからEBTを遊離する反
応になり,この変色は上の反応によるほど明瞭ではない。
c) この方法では,シアン化カリウムを加えるので通常の重金属元素は,シアノ錯イオンとなってマスキ
ングされる。マンガンは完全にはマスキングされないので,変色が不明瞭になる。この場合は,2,2',2''-
ニトリロトリエタノール(トリエタノールアミン)を加えると効果がある。
d) ナトリウムが多量(数%)に共存すると終点の判別が不明瞭になる。
10.2.2
フレーム原子吸光法
22.3によってカルシウムの濃度を,また,23.3によってマグネシウムの濃度を求め,次の式によって全
硬度を算出する。
H=2.497×CCa+4.118×CMg
ここに,
H: 全硬度(CaCO3:mg/L)
CCa: カルシウムの濃度(Ca:mg/L)
2.497: カルシウムの量を炭酸カルシウム相当量に換算する場
合の係数(100.09/40.078)
24
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
CMg: マグネシウムの濃度(Mg:mg/L)
4.118: マグネシウムの量を炭酸カルシウム相当量に換算する
場合の係数(100.09/24.305)
10.2.3
ICP発光分光分析法
22.4によってカルシウムの濃度を,また,23.4によってマグネシウムの濃度を求め,10.2.2の式によっ
て全硬度を算出する。
10.2.4
ICP質量分析法
22.5によってカルシウムの濃度を,また,23.5によってマグネシウムの濃度を求め,10.2.2の式によっ
て全硬度を算出する。
10.2.5
イオンクロマトグラフ法
22.6によってカルシウムの濃度を,また,23.6によってマグネシウムの濃度を求め,10.2.2の式によっ
て全硬度を算出する。
10.3
カルシウム硬度
10.3.1
キレート滴定法
22.2.2において滴定に要する10 mmol/L EDTA溶液の量を求め,次の式によってカルシウム硬度を算出
する。
001
.1
000
1
Ca
×
×
=
V
a
H
ここに,
HCa: カルシウム硬度(CaCO3:mg/L)
a: 22.2.2で滴定に要した10 mmol/L EDTA溶液(mL)
V: 試料(mL)
1.001: 10 mmol/L EDTA溶液1 mLの炭酸カルシウム相当量
(mg)
10.3.2
フレーム原子吸光法
22.3によってカルシウムの濃度を求め,次の式によってカルシウム硬度を算出する。
HCa=2.497×CCa
ここに,
HCa: カルシウム硬度(CaCO3:mg/L)
CCa: カルシウムの濃度(Ca:mg/L)
2.497: カルシウムの量を炭酸カルシウム相当量に換算する場
合の係数(100.09/40.078)
10.3.3
ICP発光分光分析法
22.4によってカルシウムの濃度を求め,10.3.2の式によってカルシウム硬度を算出する。
10.3.4
ICP質量分析法
22.5によってカルシウムの濃度を求め,10.3.2の式によってカルシウム硬度を算出する。
10.3.5
イオンクロマトグラフ法
22.6によってカルシウムの濃度を求め,10.3.2の式によってカルシウム硬度を算出する。
10.4
マグネシウム硬度
10.4.1
キレート滴定法
23.2によってマグネシウムの濃度を求め,次の式によってマグネシウム硬度を算出する。
HMg=4.118×CMg
ここに,
HMg: マグネシウム硬度(CaCO3:mg/L)
CMg: マグネシウムの濃度(Mg:mg/L)
25
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4.118: マグネシウムの量を炭酸カルシウム相当量に換算する
場合の係数(100.09/24.305)
10.4.2
フレーム原子吸光法
23.3によってマグネシウムの濃度を求め,10.4.1の式によってマグネシウム硬度を算出する。
10.4.3
ICP発光分光分析法
23.4によってマグネシウムの濃度を求め,10.4.1の式によってマグネシウム硬度を算出する。
10.4.4
ICP質量分析法
23.5によってマグネシウムの濃度を求め,10.4.1の式によってマグネシウム硬度を算出する。
10.4.5
イオンクロマトグラフ法
23.6によってマグネシウムの濃度を求め,10.4.1の式によってマグネシウム硬度を算出する。
11
蒸発残留物
11.1
一般事項
水中に懸濁している物質及び水を蒸発したときの残留物質を全蒸発残留物,溶解性蒸発残留物に区分し
て試験する。試験は,試料採取後,直ちに行う。直ちに行えない場合には,試料を0〜10 ℃の暗所に保存
し,できるだけ早く試験する。
a) 溶解性蒸発残留物 懸濁物質をろ別したろ液を蒸発乾固したときに残留する物質。
11.2
全蒸発残留物
試料を蒸発乾固したときに残留する物質の質量を測定して全蒸発残留物の濃度を求める。
11.2.1
器具及び装置
器具及び装置は,次による。
a) 蒸発皿 白金皿,石英ガラス皿又は磁器蒸発皿。
11.2.2
操作
操作は,次による。
a) 試料の性質によって適切な材料の蒸発皿を選び,これを105〜110 ℃の乾燥器中で約1時間加熱し,
デシケータ中で放冷した後,その質量をはかる。加熱,放冷及びひょう量を繰り返して恒量とする。
注記 例えば,試料に塩化物イオンと強力な酸化性の物質とが共存する場合には,白金皿は用いな
い。
b) 十分に振り混ぜて懸濁物質が均一になった試料を手早く採取して蒸発皿に入れる。試料は乾燥後の質
量が5 mg以上になるように採取する。試料の全量が蒸発皿に入らない場合には,数回に分けて入れ
る。試料容器に付着しやすい懸濁物質を含む場合は,適量の試料を採取して,その全量を用いて試験
する。採取した容器の器壁に付着した懸濁物質はポリスマン(ゴム管付きガラス棒)などで落として,
蒸発皿上に集める。
c) 蒸発皿の試料を蒸発乾固する。
d) この蒸発皿を105〜110 ℃で2時間加熱した後に,先のデシケータ中で放冷し,その質量をはかる。
注記 蒸発乾固には,加熱板,沸騰水浴,赤外線ランプなどを用い,蒸発中に沸騰させないように
する。また,外部からの汚染に注意する。
e) 次の式によって全蒸発残留物(mg/L)を算出する。
(
)
V
b
a
R
000
1
×
−
=
26
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ここに,
R: 全蒸発残留物(mg/L)
a: 残留物の入った蒸発皿の質量(mg)
b: 蒸発皿の質量(mg)
V: 試料(mL)
11.3
溶解性蒸発残留物
懸濁物質をろ別したろ液を蒸発乾固したときに残留する物質の質量を測定して溶解性蒸発残留物の濃度
を求める。
11.3.1
器具及び装置
器具及び装置は,次による。
a) 蒸発皿 11.2.1 a) による。
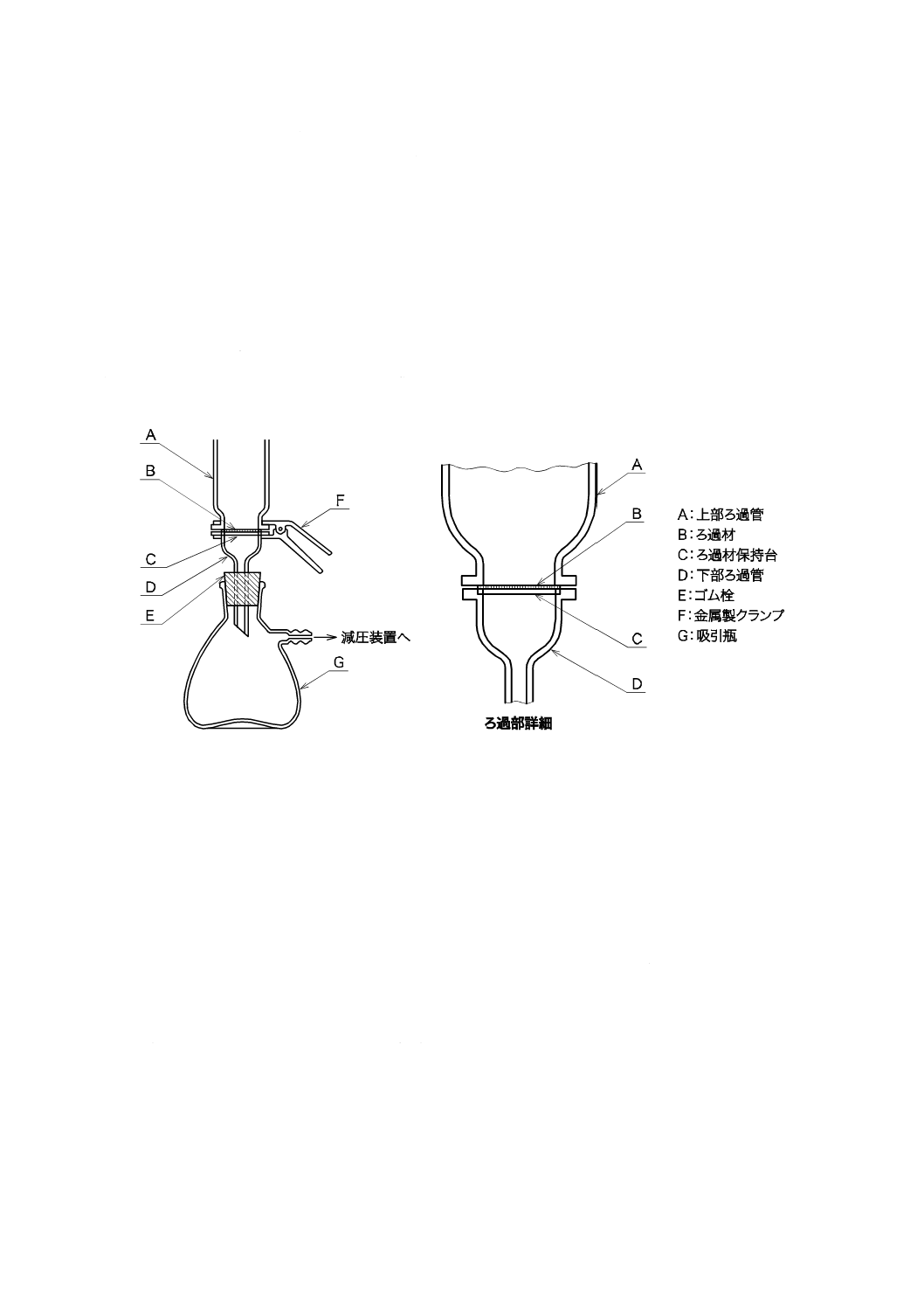
b) ろ過器(分離形) JIS K 0101の16.1 1) a)[ろ過器(分離形)]による。図6に一例を示す。
図6−ろ過器(分離形)の一例
c) ろ過材 ガラス繊維ろ紙,有機性ろ過膜又は金属性ろ過膜で,孔径1 µmで直径25〜50 mmのものを
用いる。
注記 ガラス繊維ろ紙は,目詰まりは少ないが,ガラス繊維が離脱するおそれがある。有機性ろ過
膜は,種類によって耐薬品性及び耐熱性に差があるので,取扱いに注意する。
11.3.2
操作
操作は,次による。
a) ろ過材をろ過器に取り付け,試料をろ過する。ガラス繊維ろ紙を用いる場合は,あらかじめろ過器に
取り付け,水で十分に吸引洗浄する。アクリル樹脂などのバインダ処理を行ったガラス繊維ろ紙,有
機性ろ過膜及び金属性ろ過膜は洗浄しなくてよい。
b) ろ液について11.2.2のa)〜e) に準じて操作する。
12
有機体炭素(TOC)
12.1
一般事項
有機体炭素とは,水中に存在する有機物中の炭素をいう。その定量には,燃焼酸化−赤外線式TOC分析
27
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
法,燃焼酸化−赤外線式TOCプロセス用分析計による測定方法,湿式酸化−赤外線式TOC分析法又は湿
式酸化−赤外線式プロセス用分析計による測定方法を適用する。TOC分析装置で有機体炭素を二酸化炭素
とする方式には,燃焼酸化法,高温湿式酸化法,紫外線湿式酸化法,及び光触媒湿式酸化法がある。生成
した二酸化炭素の定量には,赤外線分析法,熱伝導度測定法,及び電気伝導率測定法があるが,あらかじ
め検出率を確認しておく。
この試験は,試料採取後,直ちに行う。直ちに行えない場合には,5.4.2 d) によって保存し,できるだ
け早く試験する。
試料中に元素状態で存在する炭素の粒子(すす),炭化物,シアン化物イオン,シアン酸イオン及びチオ
シアン酸イオンが存在する場合には,有機体炭素として定量される。
全炭素(TC),無機体炭素(TIC)及び有機体炭素(TOC)の定義は,次による。
a) 全炭素(TC) 水中に存在する有機的に結合した炭素と,無機的に結合した炭素(元素状の炭素を含
む。)との合量。
b) 無機体炭素(TIC) 水中に存在する無機体の炭素の合量。すなわち,元素状,全二酸化炭素,一酸
化炭素,シアン化物イオン,シアン酸イオン及びチオシアン酸イオン中の炭素の合量。
なお,TOC分析計で,TICを二酸化炭素として測定する場合,そのほとんどは炭酸水素イオン及び
炭酸イオンに起因する。
c) 有機体炭素(TOC) 水中に存在する有機的に結合した炭素の合量。すなわち,溶存及び懸濁状で存
在する物質中の有機的に結合した炭素,シアン酸イオン,チオシアン酸イオン中の炭素の合量。
12.2
燃焼酸化−赤外線式TOC分析法
少量の試料を二酸化炭素を除去した空気又は酸素とともに高温の全炭素測定管に送り込み,有機物中の
炭素及び無機物[無機体炭素(主として炭酸塩類)]中の炭素を二酸化炭素とした後,その濃度を非分散形
赤外線ガス分析計で測定して全炭素(TC)の濃度を求める。
別に,試料を有機物が分解されない温度に保った無機体炭素測定管に送り込み,生成した二酸化炭素を
測定し,無機体炭素(TIC)の濃度を求める。
全炭素の濃度から無機体炭素の濃度を差し引いて有機体炭素(TOC)の濃度を算出する。
定量範囲:C:0.5〜25 mg/L,繰返し精度:3〜10 %(装置及び測定条件によって異なる。)
12.2.1
試薬
試薬は,次による。
a) 水 JIS K 0557で規定するA3又はA4の水による。精製した水は,容器に入れて保存すると徐々に汚
染されてTOCの濃度が高くなることがあるので,精製後は早く使用することが望ましい。
b) TOC標準液(C:1 000 mg/L) JIS K 8005で規定する容量分析用標準物質のフタル酸水素カリウム
を120 ℃で約1時間加熱し,デシケータ中で放冷する。その2.125 gをとり,少量の水に溶かして全
量フラスコ1 000 mLに移し入れ,水を標線まで加える。
c) TOC標準液(C:100 mg/L) TOC標準液(C:1 000 mg/L)10 mLを全量フラスコ100 mLにとり,
水を標線まで加える。
d) 無機体炭素標準液(C:1 000 mg/L) JIS K 8622で規定する炭酸水素ナトリウムをデシケータ中で約
3時間放置し,その3.497 gをとる。別に,JIS K 8005で規定する容量分析用標準物質の炭酸ナトリウ
ムを,あらかじめ600 ℃で約1時間加熱し,デシケータ中で放冷し,その4.412 gをとる。両者を少
量の水に溶かして全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
e) 無機体炭素標準液(C:100 mg/L) 無機体炭素標準液(C:1 000 mg/L)10 mLを全量フラスコ100 mL
28
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
にとり,水を標線まで加える。
f)
全炭素測定管 全炭素定量用触媒を充塡したもの。
g) 無機体炭素測定管 無機体炭素定量用触媒を充塡したもの。
h) キャリヤガス 二酸化炭素を除去した空気又はJIS K 1101で規定する酸素。
12.2.2
器具及び装置
器具及び装置は,次による。
a) マイクロシリンジ 20〜150 μL又は自動注入装置。
b) TOC分析装置
c) ホモジナイザ又はミキサ 分散した物質の均質化に十分な能力をもつもの。超音波装置,マグネチッ
クスターラなど。
12.2.3
準備操作
準備操作は,次による。
a) TOC分析装置を作動できる状態にする。
b) TOC標準液(C:1 000 mg/L)又はTOC標準液(C:100 mg/L)の一定量(例えば,20 μL)をマイク
ロシリンジでTOC分析装置の全炭素測定管に注入し,指示値(ピーク高さ又はピーク面積)を読み取
る。ただし,試料の炭素の濃度が低い場合には,各標準液の注入量は100〜150 μLとし,炭素の濃度
が高い場合には,注入量を少なくするか,又は一定の倍数に薄める。
c) b) の操作を5〜7回繰り返して指示値が一定になることを確かめる。
d) 試料をよく振り混ぜて均一にした後,b) と同量をマイクロシリンジで全炭素測定管に注入して,指示
値を読み取り,b) と比較して試料中の概略の全炭素の濃度(C:mg/L)を求める。
12.2.4
操作
操作は,次による。
a) 試料に懸濁物が含まれている場合には,ホモジナイザ又はミキサでよくかき混ぜてこれらを均一に分
散させる。
b) 試料の一定量[例えば,12.2.3 b) と同量]をマイクロシリンジで全炭素測定管に注入し,指示値を読
み取る。
c) 試料の一定量[例えば,12.2.3 b) と同量]をマイクロシリンジで無機体炭素測定管に注入し,指示値
を読み取る。
d) 試料を薄めた場合には,b) 及びc) の空試験としてそれぞれ同量の水をマイクロシリンジでとり,b)
及びc) の操作を行って試料について得た結果を補正する。
e) あらかじめ,次によって作成した全炭素及び無機体炭素の検量線から注入した試料中の全炭素及び無
機体炭素の濃度を求め,それぞれの濃度(C:mg/L)を算出する。
1) 12.2.3 d) で求めた試料の概略の炭素の濃度がほぼ中央になるようにTOC標準液(C:1 000 mg/L)
又はTOC標準液(C:100 mg/L)を全量フラスコ100 mLに段階的にとり,水を標線まで加える。
2) 1) で調製したTOC標準液の最高濃度のものの一定量[例えば,12.2.3 b) と同量]をマイクロシリ
ンジで全炭素測定管に注入して指示値が最大目盛値の約80 %になるようにTOC分析装置の感度及
び標準液の注入量を調節する。
3) 1) で調製した各濃度のTOC標準液の一定量[2) で定めた量]を,順次,マイクロシリンジで全炭
素測定管に注入して指示値を読み取る。
4) 空試験として,3) と同量の水をマイクロシリンジで全炭素測定管に注入して,指示値を読み取り,
29
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3) の結果を補正し,有機体炭素の濃度と指示値との関係線を作成して,これを全炭素の検量線とす
る。
5) 無機体炭素標準液(C:1 000 mg/L)又は無機体炭素標準液(C:100 mg/L)を用いて,1) で段階的
に調製したTOC標準液と同濃度の炭素を含むように無機体炭素標準液を段階的に調製する。
6) 5) で調製した各濃度の無機体炭素標準液の一定量[2) で定めた量]を,順次,マイクロシリンジ
で無機体炭素測定管に注入し,指示値を読み取る。
7) 空試験として6) と同量の水をマイクロシリンジで無機体炭素測定管に注入して,指示値を読み取
り,6) の結果を補正し,無機体炭素の濃度と指示値との関係線を作成して,これを無機体炭素の検
量線とする。
f)
次の式によって試料の全有機体炭素(TOC)の濃度(C mg/L)を算出する。
TOC=(Ct−Ci)×d
ここに,
TOC: 有機体炭素(C mg/L)
Ct: 注入試料中の全炭素(C mg/L)
Ci: 注入試料中の無機体炭素(C mg/L)
d: 注入試料の希釈倍数
g) 次のいずれかの溶液を調製し,測定範囲の80 %近くなるように希釈して測定し,TOCの検出率を確
認する。
1) 酒石酸溶液(C:100 mg/L) JIS K 8532で規定するL(+)-酒石酸をJIS K 8228で規定する過塩素
酸マグネシウム(乾燥用)を入れたデシケータ中で18時間以上放置し,その0.312 5 gをとり,水
に溶かして全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
2) 1,10-フェナントロリン酸溶液(C:100 mg/L) JIS K 8789で規定する1,10-フェナントロリン一水
和物0.137 6 gをとり,水に溶かして全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
3) L-グルタミン酸溶液(C:100 mg/L) JIS K 9047で規定するL-グルタミン酸を約80 ℃で約3時
間乾燥し,デシケータ中で放冷し,その0.245 gをとり,水に溶かして全量フラスコ1 000 mLに移
し入れ,水を標線まで加える。
4) 2-プロパノール溶液(C:100 mg/L) 全量フラスコ50 mLに水約30 mLを入れ,密栓してその質
量を測定する。これにJIS K 8839で規定する2-プロパノール(イソプロピルアルコール)の約10.6
mLを速やかに加えて密栓し,その質量を測定する。次いで水を標線まで加える。この溶液の濃度
は,前後の質量の差から求める[2-プロパノール1 gは,炭素(C)0.599 gに相当する。]。この溶液
1 mLを全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
12.2.5
留意事項
a) この方法では,有機体炭素が少なく無機体炭素の多い試料では,誤差が大きくなる。
b) 無機体炭素の多い試料では,あらかじめ試料に塩酸を加えてpH 2以下にし,JIS K 1107で規定する窒
素2級を通気して無機体炭素を除去した後,その少量を高温の全炭素測定管に送り込み,炭素の定量
を行ってこれを有機体炭素の量とする方法がある。その方法は,有機体炭素に比べて無機体炭素が多
い試料の場合に優れている。ただし,揮発性の有機物を含む場合には誤差が大きい。
12.3
TOCプロセス用分析計(燃焼酸化−赤外線式)による測定方法
計測器に供給した試料に酸を加えてpHを2以下にし,通気して無機体炭素を除去した後,その一定量
をキャリヤガスとともに高温の全炭素測定管に送り込み,有機物中の炭素を二酸化炭素とし,その濃度を
非分散形赤外線ガス分析計で測定して有機体炭素(TOC)の濃度を求める。
30
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
定量範囲:C:0.05〜1 000 mg/L,繰返し精度:3〜10 %(装置及び測定条件によって異なる。)
12.3.1
試薬
試薬は,次による。
a) 水 12.2.1 a) による。
b) TOC標準液(C:1 000 mg/L) 12.2.1 b) による。
c) TOC標準液(C:100 mg/L) 12.2.1 c) による。
d) ゼロ校正液 a) の水を用いる。
e) スパン校正液 TOC標準液(C:100 mg/L)[又はTOC標準液(C:1 000 mg/L)]の適量を全量フラ
スコにとり,水を標線まで加える。計測器の測定範囲の約80 %に相当するTOCの濃度になるように
調製する。TOC標準液を使用時に調製する。
f)
酸溶液 JIS K 9005で規定するりん酸,JIS K 8180で規定する塩酸又はJIS K 8951で規定する硫酸で
TOCの濃度のできるだけ少ないものを用い,所定の濃度に調製する。
g) キャリヤガス 12.2.1 h) による。
12.3.2
器具及び装置
器具及び装置は,次による。
a) TOCプロセス用分析計 JIS K 0805で規定する超純水用(最大目盛値が1 000 μg/L以下)の燃焼酸化
−赤外線式TOCプロセス用分析計。
12.3.3
準備操作
準備操作は,次による。
a) 酸溶液及びキャリヤガスを,計測器に供給する。
b) 計測器の電源を入れ,各部の機能及び指示記録部を安定させる。
c) ゼロ校正液及びスパン校正液を用いて計測器を校正する。
12.3.4
操作
操作は,次による。
a) 試料を,計測器に供給して指示値が安定したことを確認する。
b) 指示値から試料中の有機体炭素(TOC)の濃度(C:mg/L)を求める。
12.4
湿式酸化−赤外線式TOC分析法
あらかじめ試料に酸を加えてpHを2以下にし,通気して無機体炭素を除去する。無機体炭素を除去し
た試料の一定量をペルオキソ二硫酸塩及びキャリヤガスとともに,湿式酸化反応器に送り込み,有機物中
の炭素を二酸化炭素とした後,その濃度を非分散形赤外線ガス分析計で測定してTOC濃度を求める。この
方法は,TOCの濃度に比べて無機体炭素が多い場合は,誤差が少ない。ただし,揮発性の有機物を含む場
合には,誤差が多い。
定量範囲:C:0.01〜1 mg/L,繰返し精度:3〜10 %(装置及び測定条件によって異なる。)
12.4.1
試薬
試薬は,次による。
a) 水 JIS K 0557で規定するA4の水で,4.7 a) の二酸化炭素を含まない水を用いるか,又は同等の質
の水を用いる。あらかじめ空試験を行い,使用の可否を確認する。超純水製造装置で調製した水を用
いてもよい。
b) 酸溶液 JIS K 8951で規定する硫酸又はJIS K 9005で規定するりん酸で有機物のできるだけ低いもの
を用い,所定の濃度に調製する。
31
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
c) ペルオキソ二硫酸塩溶液 JIS K 8253で規定するペルオキソ二硫酸カリウム又はペルオキソ二硫酸ナ
トリウムで有機物のできるだけ低いものを用い,所定の濃度に調製する。
d) キャリヤガス 12.2.1 h) による。
e) TOC標準液(C:100 mg/L) 12.2.1 c) による。
f)
TOC標準液(C:1 mg/L) TOC標準液(100 mg/L)5 mLを全量フラスコ500 mLにとり,水を標線
まで加える。使用時に調製する。
12.4.2
器具及び装置
器具及び装置は,次による。
a) マイクロシリンジ 0.2〜20 mL又は自動注入装置。
b) TOC分析装置 最大目盛値が1 000 μg/L以下の湿式酸化−赤外線式TOC分析装置。
12.4.3
準備操作
準備操作は,次による。
a) TOC分析装置を作動できる状態にする。
b) TOC標準液(C:1 mg/L)の一定量(例えば,20 mL)をマイクロシリンジでTOC分析装置に注入し,
指示値(ピーク高さ又はピーク面積)を読み取る。このときの一定量とは,試料の全炭素の濃度又は
無機体炭素の濃度の予想値によって,適切な指示値が得られる注入量をさす。この操作を繰り返し指
示値が一定になることを確かめる。
c) 試料の適量を適切な容量の細口共栓瓶(例えば,容量500 mL)に入れ(上部に空間を残す),酸溶液
を添加してpHを2以下にした後,キャリヤガスを通気して無機体炭素を除去する。無機体炭素の除
去には,酸溶液の量,通気するガスの流量,通気時間,処理する試料の量,処理容器の構造などが影
響するため,12.2.1 e) によって調製した無機体炭素標準液(C:100 mg/L)を薄め,調製した無機体
炭素の標準液(例えばC:1 mg/L)を用いて除去が十分に行われていることを確認する。
d) 無機体炭素を除去した試料の一定量[例えば,b) と同量]をマイクロシリンジで注入し,指示値を読
み取り,b) の指示値と比較して概略のTOCの濃度(C:μg/L)を求める。
12.4.4
操作
操作は,次による。
a) 12.4.3 c) で無機体炭素を除去した試料の一定量[12.4.3 d) と同量]をマイクロシリンジでTOC分析
装置に注入し,指示値を読み取る。
b) あらかじめ,次によって作成したTOCの検量線から試料中のTOCの濃度(C:μg/L)を求める。
1) 12.4.3 d) で求めた試料の概略のTOCの濃度(C:μg/L)がほぼ中央になるようにTOC標準液(C:
1 mg/L)を全量フラスコ100 mLに段階的にとり,水を標線まで加える。
2) 1) で調製したTOC標準液の最高濃度のものの一定量[例えば,12.4.3 d) と同量]をマイクロシリ
ンジでTOC分析装置に注入し,指示値が最大目盛値の約80 %になるようにTOC分析装置の感度及
び標準液の注入量を調節する。
3) 順次,1) で調製した各濃度のTOC標準液の一定量[2) で定めた量]をマイクロシリンジでTOC
分析装置に注入し,指示値を読み取る。
4) 空試験として2) と同量の水をマイクロシリンジでTOC分析装置に注入し,指示値を読み取る。3) で
得た結果を補正し,TOCの濃度(C:μg/L)と指示値との関係線を作成し,これをTOCの検量線と
する。
12.4.5
留意事項
32
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) 紫外線を照射して有機体炭素を二酸化炭素とする紫外線湿式酸化法では,用いる水銀灯の紫外線の放
射強度が徐々に低下するので,適切な時期に交換する。
b) 試料に揮発性の有機物が含まれる場合には,12.4.3 c) の操作において通気することによって試料から
失われることがあるため,通気量は必要以上に大きくしない。
c) 無機体炭素を除去する際に,使用する容器及びその材料などによって試料が汚染される割合をできる
だけ少なくするためには,できるだけ多量の試料について無機体炭素の除去操作を行い,そのうち一
部の試料を用いて測定するとよい。
12.5
TOCプロセス用分析計(湿式酸化−赤外線式)による測定方法
計測器に供給した試料に酸を加えてpHを2以下にし,通気して無機体炭素を除去した後,その一定量
(又は一定流量)をペルオキソ二硫酸塩カリウム及びキャリヤガスとともに湿式酸化反応器に送り込み,有
機物中の炭素を二酸化炭素とし,その濃度を非分散形赤外線ガス分析計で測定して有機体炭素(TOC)の
濃度を求める。
定量範囲:C:0.01〜1 mg/L,繰返し精度:3〜10 %(装置及び測定条件によって異なる。)
12.5.1
試薬
試薬は,次による。
a) 水 12.4.1 a) による。
b) 酸溶液 12.4.1 b) による。
c) ペルオキソ二硫酸塩溶液 12.4.1 c) による。
d) キャリヤガス 12.2.1 h) による。
e) TOC標準液(C:100 mg/L) 12.2.1 c) による。
f)
ゼロ校正液 a) の水を用いる。
g) スパン校正液 12.3.1 e) による。
12.5.2
器具及び装置
器具及び装置は,次による。
a) TOCプロセス用分析計 JIS K 0805で規定する超純水用(最大目盛値が1 000 μg/L以下)の湿式酸化
−赤外線式TOCプロセス用分析計。
12.5.3
準備操作
準備操作は,次による。
a) 酸溶液,ペルオキソ二硫酸塩溶液及びキャリヤガスを,計測器に供給する。
b) 計測器を作動し,各部の機能及び指示記録部を安定させる。
c) ゼロ校正液及びスパン校正液を用いて,計測器を校正する。
12.5.4
操作
操作は,次による。
a) 試料を計測器に供給して指示値が安定したことを確認する。
b) 指示値から試料中にTOC(C:μg/L)を求める。
13
ヘキサン抽出物質
13.1
一般事項
ヘキサン(n-ヘキサン)抽出物質とは,試料を微酸性とし,ヘキサン抽出を行った後,約80 ℃でヘキ
サンを揮散させたときに残留する物質をいう。
33
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
この試験は,主として揮散しにくい鉱物油及び動植物油脂類の定量を目的とするが,これらのほかヘキ
サンに抽出された揮散しにくいもの(例えば,炭化水素誘導体,脂肪酸類,エステル類,アミン類,フェ
ノール類,界面活性剤,コロイド状硫黄など)は,定量値に含まれる。この試験には,抽出法を適用する。
13.2
試料採取
試料の採取は,次による。
13.2.1
試薬
試薬は,次による。
a) 塩酸(1+1) JIS K 8180で規定する塩酸を用いて調製する。
b) メチルオレンジ溶液(1 g/L) JIS K 8893で規定するメチルオレンジ0.1 gを熱水100 mLに溶かす。
13.2.2
器具及び装置
器具及び装置は,次による。
a) 試料容器 共栓広口ガラス瓶(又は共栓ガラス瓶)1〜2 Lのもの。使用前にヘキサンでよく洗ってお
く。
13.2.3
試料採取方法
試料採取方法は,次による。
a) 通水状態の配管装置などからの採取 配管,装置などが通水状態の場合には,試料採取弁を開き,試
料採取配管内に滞留している水の約5倍量を約1 L/minの割合で流出させてから試料容器に受け,適
切な空間が残る程度に採取をとどめる。
なお,試料を採水する際に,試料容器を試料で洗わない。また,試料採取直前に流量を変更しては
ならない。
b) 高温高圧状態にある配管・装置などからの採取 高温水の場合は,冷却器を試料採取管に設けて室温
以下に冷却し,a) に準じて採取する。高圧水(圧力が1.96 MPa以上)の場合には,減圧器を設けて
減圧した後に採取し,高温であれば冷却器を通して室温以下に冷却する。
c) 負圧状態にある配管・装置などからの採取 負圧水の場合には,昇圧器で大気圧にしてからa) に準
じて採取する。
なお,負圧水で高温の場合は,昇圧器の前に冷却器を設けて室温にしてから大気圧にする[JIS K
0094の4.3(採水弁を用いる採取)を参照]。
注記 装置などが停止状態にあるときは,油状物質が配管及び装置中で水と分離していることが多
いため,通水速度及び通水時間によって油状物質の濃度に変動が生じる。試料採取弁及び配
管中に油状物質が付着しているおそれがある場合には,試料採取弁を全開して約10分間通水
してから,約1 L/minの割合で,更に10分間通水する。この操作を繰り返して洗浄する。
13.2.4
試料の取扱い
試料の取扱いは,次による。
a) 13.2.3によって採取した試料は,他の容器に移し替えたり,一部を採取したりしてはならない。試験
には全量を用いる。
b) 試料の量は,試料を入れた容器の質量から試料容器の質量を差し引いて求めるか,又は試料を採取し
たときに試料容器の水面の位置に印を付けておき,試験終了時に印のところまで水を入れてその水の
体積を試料の量とする。
c) 試料を保存したり,運搬したりする必要がある場合には,指示薬としてメチルオレンジ溶液(1 g/L)
5〜7滴を加え,溶液の色が赤くなるまで塩酸(1+1)を加えて密栓する。
34
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
なお,油状物質が浮上している場合には,運搬中の振動でにじみやすいので,試料容器のすり合わ
せに注意する。
13.3
抽出法
試料をpH 4以下の塩酸酸性にして,ヘキサンで抽出を行い,80 ℃でヘキサンを揮散させて残留する物
質の質量を測定してヘキサン抽出物質を定量する。
定量範囲:5〜500 mg/L(試料量1 Lの場合),繰返し精度:10〜20 %
13.3.1
試薬
試薬は,次による。
a) 水 JIS K 0557で規定するA3の水。
b) 塩酸(1+1) 13.2.1 a) による。
c) 硫酸ナトリウム JIS K 8987で規定するもの。
d) メチルオレンジ溶液(1 g/L) 13.2.1 b) による。
e) ヘキサン JIS K 8848で規定するもの。
f)
窒素 JIS K 1107で規定する窒素2級。
13.3.2
器具及び装置
器具及び装置は,次による。
a) 分液漏斗 200 mL及び1 000〜3 000 mL(試料の量に応じた適切な大きさのもの)で脚部の短いもの。
使用前にヘキサンで洗う。コックにワセリンなどの滑剤を塗布しない。
b) 乾燥器 80±5 ℃に温度調節できるもの。
c) 加熱板又はマントルヒータ 80±5 ℃に温度調節できるもの。温度調節ができる水浴を用いてもよい。
d) 蒸留装置 共通すり合わせで,蒸留フラスコ(容量50〜100 mL),トの字形連結管及びリービッヒ冷
却器(長さ300 mm)を接続できるもの。いずれも使用前にヘキサンでよく洗っておく。
e) 蒸発容器 アルミニウムはく皿,白金皿又はビーカ。容量50〜100 mLで,できるだけ質量の小さい
もの。いずれも使用前にヘキサンでよく洗い,80±5 ℃で約30分間加熱し,デシケータ中で放冷した
後,質量を0.1 mgの桁まで求めておく。
13.3.3
操作
操作は,次による。
a) 13.2.3で採取した試料の全量(ヘキサン抽出物を5〜500 mg/L含む。)を分液漏斗1 000〜3 000 mLに
移し,指示薬としてメチルオレンジ溶液(1 g/L)2,3滴を加え,溶液の色が赤に変わるまで塩酸(1
+1)を滴加する。
注記 ヘキサン抽出物質の質量が5 mg以下で定量が困難な場合には,JIS K 0102の24.3(抽出容器
による抽出法)又は24.4(捕集濃縮・抽出法)によって試験するとよい。
b) 試料容器を約20 mLずつのヘキサンで2回洗い,洗液を分液漏斗1 000〜3 000 mLに加える。約2分
間激しく振り混ぜ,放置する。試料の性質によって,エマルションが生成したり,ヘキサン層が濁っ
たりした場合は13.3.4の方法による。
c) 水層は試料容器に戻し,更に分液漏斗1 000〜3 000 mLを静かに揺り動かして,残った水層をできる
だけ分離して試料容器に戻す。ヘキサン層は分液漏斗200 mLに移す。この分離操作は,分離する水
層が1 mL以下になるまで続ける。試料が多量のグリース類又は固体油脂を含む場合には,水層を分
離する前にヘキサンを追加する。
d) 試料容器の水層をa) で使用した分液漏斗1 000〜3 000 mLに入れ,再び,b) 及びc) の操作を行って
35
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ヘキサン層と水層とを分離し,ヘキサン層をc) の分液漏斗200 mLに加える。
e) 分液漏斗1 000〜3 000 mLを少量のヘキサンで洗い,洗液を分液漏斗200 mLに加える。
f)
分液漏斗200 mLを静かに揺り動かして静置し,ヘキサンを損失しないように注意しながら混入した
水分を十分に分離除去する。この分離操作は,分離する水層が1 mL以下になるまで続ける。
g) ヘキサン層に水約20 mLを加え,約1分間振り混ぜて放置し,水層を捨てる。この洗浄操作を洗液が
メチルオレンジに対して黄色になるまで数回繰り返す。できるだけ水層を除去する。
h) ヘキサン層が濁っている場合には,水層をできるだけ分離した後,ヘキサン層に硫酸ナトリウム3〜5
gを加えて振り混ぜ,水分を除く。硫酸ナトリウムよりも,JIS K 8150で規定する塩化ナトリウム又
はJIS K 8960で規定する硫酸アンモニウムを使用する方が効果的な場合もある。ただし,ヘキサンに
溶ける物質を含む試薬は使用しない。
i)
分液漏斗200 mLの脚部を乾いたろ紙で拭き取り,脱脂綿又はろ紙を用いてヘキサン層をろ過し,蒸
留装置の蒸留フラスコに移し入れる。使用する脱脂綿又はろ紙は,ヘキサンで十分に洗って抽出物質
を除いたものを使用し,ろ過の際にはあらかじめ少量のヘキサンで潤しておく。ろ過したヘキサン層
が蒸留フラスコに一度に入りきらないときは,2,3回に分割してヘキサンを留出させる。
j)
分液漏斗200 mLを少量のヘキサンで洗い,この洗液もi) と同じ操作でろ過し,蒸留装置の蒸留フラ
スコに移し入れる。使用した脱脂綿又はろ紙は,ヘキサン約5 mLずつで2回洗い,この洗液も蒸留
フラスコに移し入れる。
k) 蒸留フラスコをマントルヒータに入れ,トの字形連結管及びリービッヒ冷却器を接続して,マントル
ヒータの温度を約80 ℃に調節し,ヘキサンを毎秒1滴の留出速度で蒸留し,留出するヘキサンを受
器に受ける。蒸留は,蒸留フラスコ中の液量が約2 mLになるまで蒸留を続ける。トの字形連結管の
上部口から窒素を室温になるまで送入する。
l)
蒸留フラスコ中の残留液を質量既知の蒸発容器に移し入れる。蒸留フラスコを少量のヘキサンで3回
洗い,この洗液も蒸発容器に加える。蒸発容器を約80 ℃に保った加熱板の上又はマントルヒータの
中に置いてヘキサンを揮散させる。ヘキサンを揮散後,蒸発容器中に水分が認められる場合には,JIS
K 8034で規定するアセトンを加えて蒸発を繰り返し,水分を除去する。水分中に塩類が残留すると誤
差になるので注意する。塩類が残留する場合にはm) の操作を行い,ヘキサン抽出物質の質量を求め
た後,ヘキサン抽出物質を少量のヘキサンを加えて溶かし分離する。この操作を繰り返し行い,ヘキ
サン抽出物質を除去した後,m) の操作を行って残留物質の質量を求めて補正する。
注記 ヘキサンを揮散させる際,ヘキサンに引火しないように十分に注意する。また,溶媒は揮散
廃棄せずに,できるだけ回収する。
m) 蒸発容器の外側を湿った清浄な布などで拭い,次に,乾いた清浄な布などでよく拭って,80±5 ℃に
調節した乾燥器に入れ,約30分間加熱する。蒸発容器をデシケータ中で約30分間放冷した後,その
質量を0.1 mgの桁まではかり,蒸発容器の質量を差し引き,ヘキサン抽出物質の質量(mg)を求め
る。
n) 空試験として,この試験に使用した全ヘキサンと同量のヘキサンを蒸留フラスコにとり,k)〜m) の
操作を行って,残留物質の質量(mg)を求める。蒸留フラスコにヘキサンが入りきらないときは,2,
3回に分割して留出させる。
o) 次の式によって試料中のヘキサン抽出物質の濃度(mg/L)を算出する。
(
)
V
b
a
P
000
1
×
−
=
36
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ここに,
P: ヘキサン抽出物質(mg/L)
a: 試験操作におけるヘキサン抽出物質の質量(mg)
b: 空試験における残留物質の質量(mg)
V: 試料(mL)
13.3.4
留意事項
エマルションが生成したり,ヘキサン層が濁ったりした場合は,次に示すa) の加熱還流法又はb) の遠
心分離法のいずれかの方法によってヘキサン層の分離を容易にすることができる。
a) 加熱還流法
1) 分液漏斗中の水層をできるだけ元の試料容器に戻す。
2) これに,JIS K 8150で規定する塩化ナトリウム又はJIS K 8960で規定する硫酸アンモニウム約10 g
(ヘキサンに溶ける物質を含まないもの)を加える。
3) 次に,分液漏斗の口に約300 mmの共通すり合わせリービッヒ冷却器又はジムロート冷却器を取り
付け,約80 ℃に保った恒温水浴中に分液漏斗を浸し,約10分間ヘキサンを還流させる。
b) 遠心分離法
1) 分液漏斗中のヘキサン層及びエマルション層にJIS K 8150で規定する塩化ナトリウム又はJIS K
8960で規定する硫酸アンモニウム約10 gを加えて振り混ぜた後,少量の水で遠心分離管に移す。
2) 回転数8 000 rpm以上で約5分間遠心分離すると,エマルション層は僅かになり,ヘキサン層の分
離を容易にすることができる。
c) a) 又はb) で調整した溶液を分液漏斗に戻し,13.3.3 c) 以下の操作に移る。
14
溶存酸素
14.1
一般事項
溶存酸素の定量には,インジゴカルミン比色法,隔膜電極法,光学式センサ法又は溶存酸素プロセス用
分析装置による測定方法を適用する。この試験は,試料採取後,直ちに行う。
14.2
インジゴカルミン比色法
インジゴカルミン比色法は,アルカリ性でインジゴカルミン[2-(1,3-ジヒドロ-3-オキソ-5-スルホ-2H-
インドール-2-イリデン)-2,3-ジヒドロ-3-オキソ-1H-インドール-5-スルホン酸二ナトリウム]とグルコース
とを加え,試料中の溶存酸素によって生じる呈色を,溶存酸素比色液と比較して定量する。この方法は,
同一色の濃淡を比較する比色法ではなく,溶存酸素の濃度がO:0〜4 µg/Lの範囲では黄色中の緑,溶存酸
素の濃度がO:4〜20 µg/Lの範囲では黄色中の赤の濃淡を判別する。
定量範囲:O:0〜60 µg/L
14.2.1
試薬
試薬は,次による。
a) インジゴカルミン溶液 JIS K 8092で規定するインジゴカルミン20 mgとJIS K 8824で規定する
D(+)-グルコース0.2 gとを水5 mLに溶かした後,JIS K 8295で規定するグリセリン75 mLを加えて
溶かす。着色瓶に入れて冷暗所に保存する。
注記 この溶液は,密栓して10 ℃以下の暗所に保存すれば約1か月間は安定である。これ以外の
場所に放置するとインジゴカルミンは急速に分解する。
b) 水酸化カリウム溶液 JIS K 8574で規定する水酸化カリウム53 gを水100 mLに溶かす。使用時に調
製する。
37
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
c) アルカリ性インジゴカルミン溶液 インジゴカルミン溶液8 mLと水酸化カリウム溶液2 mLとをよく
混合する。この溶液は,使用の約30〜40分間前に調製し,暗い赤から黄色になるまで放置した後,使
用する。2時間以上経過したものは使用しない。
なお,溶液の色が暗い赤から完全に黄色(レモン色)になるまでの時間は,グリセリンの品質及び
温度によって一定しない。変色に要する時間をあらかじめ確認し,完全に黄色(レモン色)になって
から使用する。温度が低いと時間を要する。
d) 塩化鉄(III)溶液 JIS K 8142で規定する塩化鉄(III)六水和物45.1 gとJIS K 8180で規定する塩酸
10 mLとを水約300 mLに溶かし,水で1 Lとする。
e) 塩化コバルト(II)溶液 JIS K 8129で規定する塩化コバルト(II)六水和物59.3 gとJIS K 8180で
規定する塩酸10 mLとを水約300 mLに溶かし,水で1 Lとする。
f)
硫酸銅(II)溶液 JIS K 8983で規定する硫酸銅(II)五水和物62.5 gとJIS K 8180で規定する塩酸
10 mLとを水約300 mLに溶かし,水で1 Lとする。
g) 溶存酸素比色液 溶存酸素の濃度に応じて塩化鉄(III)溶液,塩化コバルト(II)溶液及び硫酸銅(II)
溶液を表6に示す割合で比色瓶にとり,JIS K 8180で規定する塩酸1 mLを加えて水を100 mLの標線
まで加える。調製後2時間以上経過すると,比色液の色調が変わることがある。変色したものは調製
し直す。
表6−溶存酸素比色液
溶存酸素
O:µg/L
塩化鉄(III)溶液
mL
塩化コバルト(II)溶液
mL
硫酸銅(II)溶液
mL
0
11.67
0.25
−
2
8.75
0.75
−
4
7.20
1.35
−
6
6.05
1.70
−
8
5.05
1.95
−
10
4.17
2.08
−
15
3.33
3.13
−
20
2.13
4.33
−
25
1.27
4.80
−
30
1.10
4.87
0.07
35
0.97
5.03
0.37
40
0.80
5.17
0.73
45
0.67
5.37
0.93
50
0.57
6.10
2.70
55
0.47
7.23
4.37
60
0.33
8.33
6.00
14.2.2
器具及び装置
器具及び装置は,次による。
a) 比色瓶 図7に示すように,内径約40 mmの丸底で,容量100 mL,ガラスに色がないもの。
b) 試料採取器 図8に示すように,内径約40 mmで,容量100±5 mL,ガラスに色がないもの。
注記 試料採取器は,できるだけ容量の等しいものを2個ずつ組み合わせて用いるほうが誤差が少
なくなる。また,ガラスの肉厚,外径は比色瓶とできるだけ等しくすると比色しやすい。
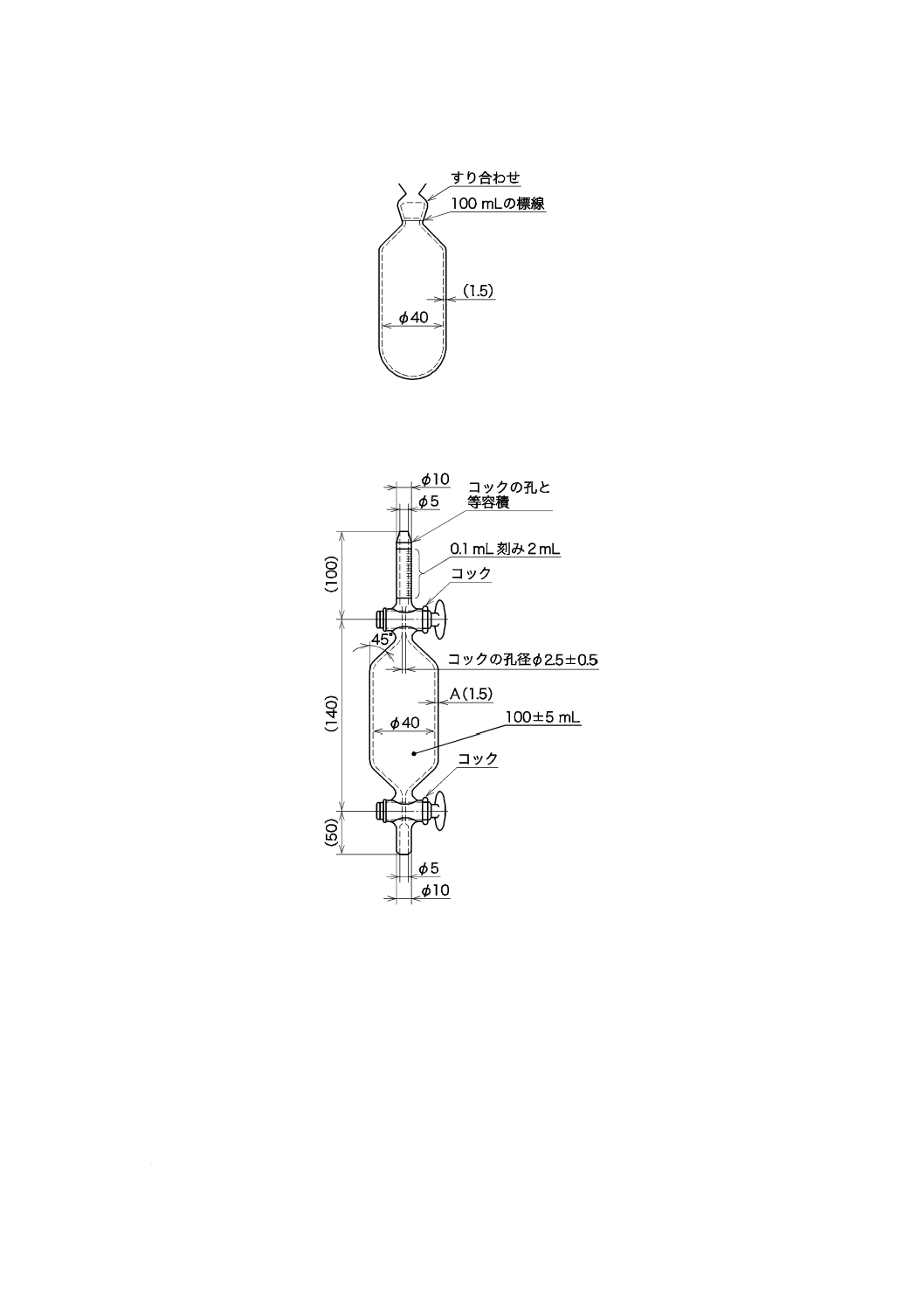
38
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
単位 mm
図7−比色瓶の一例
単位 mm
図8−試料採取器の一例
14.2.3
試料採取
試料採取は,次による。
a) 試料採取器の入口を試料採取口より高い位置に支持するように設置し,試料採取器の下端を軟質塩化
ビニル管で試料採取口に接続する。試料の温度が室温より高い場合には,試料の温度が室温より1〜
2 ℃低くなるように試料配管中に適切な冷却器を設ける。
1) 試料採取口と試料採取器とを接続する導管は,空気の浸透しにくい材質を選び,できるだけ短くし
て使用する。
注記 通常,使用する各種の導管のうち空気の浸透の少ないものは,軟質塩化ビニル管,硬質ポリ
39
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
エチレン管,ナイロン管などである。
2) 冷却器に代え,冷却蛇管を使用する場合には,冷却水調節用弁を冷却蛇管の入口に設けて冷却水を
流してあふれさせ,試料の流量調節弁は冷却蛇管の出口に設ける。
b) 試料採取器に試料が8〜12秒間で満たされるように,試料の流量を調節し,試料配管中の滞留水(元
の試料)が完全に入れ替わるように,連続して試料を十分に流す。
なお,試料配管を断続的に使用する場合にも,試料配管及び冷却蛇管中の滞流水(元の試料)を完
全に置換するのに必要な時間だけ試料を流す。
c) 試料採取器の上部のコックを閉じ,直ちに下部のコックを閉じ,接続配管を外す。試料採取器を逆さ
にして気泡が全くないことを確かめた後,試料とする。ただし,少しでも気泡があれば,試料をとり
直す。
14.2.4
操作
操作は,次による。
a) アルカリ性インジゴカルミン溶液を試料採取器の試薬注入部の最上部の基線まで満たし,試薬注入部
のコックを開き,下部のコックで調節しながらアルカリ性インジゴカルミン溶液1.2 mLを添加する。
b) 両端のコックを閉じた後,試料採取器を約1分間繰り返し転倒させて十分に混ぜ合わせ,約5分間放
置する。
c) その呈色を溶存酸素比色液と比較して,該当する溶存酸素比色液を求め,相当する溶存酸素の濃度
(O:µg/L)を求める。比色時の光源は,一般に昼光とし,直射日光を避ける。
注記 アルカリ性インジゴカルミン溶液を試料採取器の試薬注入部に満たす際,試薬注入部に付着
した水をろ紙などで完全に除去する必要はない。むしろ微量の水が残っている方が試薬注入
時に試薬注入部の底部に気泡を形成しない。この微量の水と反応した試薬の部分はすぐに浮
上するので,試薬1.5〜2 mLを試薬注入部に満たしておけば,変色した試薬の部分は試料採
取器内には入らず,測定値は影響を受けない。
14.2.5
留意事項
この試験方法には,次に示す妨害物質などがあり,その対応も含めて示す。
a) ヒドラジニウムイオン(ヒドラジン),亜硫酸イオン及びタンニンは,それぞれ1 mg/Lまでは妨害し
ない。鉄(III),シクロヘキシルアミン及びモルホリン(テトラヒドロキシ-1,4-オキサジン)は,それ
ぞれ4 mg/Lまでは許容できる。
b) 鉄(II)Fe:20 µg/L以上及び銅(II)Cu:20 µg/L以上は妨害する。
c) これらの妨害物質を許容量以上含有する場合には,試料を強酸性陽イオン交換樹脂(H+形)と強塩基
性陰イオン交換樹脂(I形)(OH−形)との混合物を充塡したカラムに通した後,その流出液について
試験すれば,妨害を除くことができる。
14.3
隔膜電極法
隔膜電極を試料に浸せきして溶存酸素濃度を測定する。隔膜電極は酸素透過性のある隔膜,対極,電解
液などから構成され,隔膜を透過した酸素が還元されて生じる電流を測定する。この電流が試料中の溶存
酸素濃度と比例することを利用し,定量する。
定量範囲:O:1 µg/L以上,繰返し精度:2〜10 %
なお,試験目的によって低濃度,高濃度のレンジ切替え可能な方式(例えば,0〜20 µg/L,0〜200 µg/L
など)を用いる。
14.3.1
試薬
40
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
試薬は,次による。
a) ゼロ校正液 ゼロ校正液は,次の1) 又は2) のいずれかによる。
1) JIS K 8061で規定する亜硫酸ナトリウム約1 gを水に溶かし,水を加えて500 mLとする。使用時に
調製する。
JIS K 8129で規定する塩化コバルト(II)六水和物を微量添加すると,溶存酸素は容易に亜硫酸
ナトリウムによって還元除去される。
2) 水に窒素(JIS K 1107で規定する窒素の1級)を通気して溶存酸素を除去したもの。水(A2又は
A3の水)を窒素で,ばっ気して溶存酸素を除去する方法では,ばっ気に用いる窒素中の酸素の濃度
によって除去できる濃度が決まる。例えば,JIS K 1107で規定する窒素1級を用いる場合は,この
窒素には酸素が最大限5 vol ppm(2級では,50 vol ppm)含まれることが許容されている。したが
って,この窒素でばっ気した場合の溶存酸素の残留濃度はブンゼン吸収係数を用いて計算すると
O:0.22 μg/Lとなる。すなわち,
22
.0
28
221
.0
994
413
.
22
999
.
31
5
031
.0
≒
=
×
×
=
O
ここに,
O: 溶存酸素(O:μg/L)
0.031: 20 ℃での酸素のブンゼン吸収係数(mL/mL)
5: 窒素中の酸素の濃度(5 μl/L)(5 vol ppm)
31.999: 酸素の分子量
22.413 994: 標準状態における気体のモル体積(L)
通気時間は,15〜25 ℃で水1 Lの場合に,約90分間通気する。水温を上げると通気時間は短く
なる。
3) これらの溶液は,ゼロ調節に用いる。
b) スパン校正液 スパン校正液は,次による。
1) 水酸化カリウム溶液(250 g/L)(JIS K 8574で規定する水酸化カリウムを用いて調製する。)で洗浄
した空気を,流量約1 L/minで球形又は板状のガラスろ過板G2又はG3を用いて水に通気し,溶存
酸素を飽和させる。スパン調節を行う直前に調製する。
2) 通気時間は,水200 mLの場合には5〜10分間,500 m Lの場合には10〜20分間通気する。
3) 溶存酸素飽和水は,試料の温度と±0.5 ℃で一致する温度のものを調製する。水中の飽和溶存酸素
量と水温との関係を表7に示す。
41
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表7−水中の飽和溶存酸素量と水温の関係(1 013 hPa)
水温
℃
溶存酸素
(O:mg/L)
水温
℃
溶存酸素
(O:mg/L)
水温
℃
溶存酸素
(O:mg/L)
水温
℃
溶存酸素
(O:mg/L)
0
14.62
12
10.78
24
8.42
36
6.84
1
14.22
13
10.54
25
8.26
37
6.73
2
13.83
14
10.31
26
8.11
38
6.62
3
13.46
15
10.08
27
7.97
39
6.52
4
13.11
16
9.87
28
7.83
40
6.41
5
12.77
17
9.67
29
7.69
41
6.31
6
12.45
18
9.47
30
7.56
42
6.21
7
12.14
19
9.28
31
7.43
43
6.12
8
11.84
20
9.09
32
7.31
44
6.02
9
11.56
21
8.92
33
7.18
45
5.93
10
11.29
22
8.74
34
7.07
11
11.03
23
8.58
35
6.95
14.3.2
器具及び装置
器具及び装置は,次による。
a) 溶存酸素測定容器 ガラス容器100〜300 mLにゴム栓を付け,栓に隔膜電極,温度計,試料注入管及
び排出するための排出管を取り付けた流液形のもの。このほかに,溶存酸素測定瓶,培養瓶などを用
いてもよい。図9に一例を示す。
A: ゴム栓
B: ガラス容器
C: 回転子
D: マグネチックスターラ
E: 温度計
F: 排出管
G: ガラス管
H: ピンチコック
I: 隔膜電極
図9−溶存酸素測定容器の例
b) 校正用容器 隔膜電極を所定位置まで挿入できるもの。a) の溶存酸素測定容器を用いてもよい。
c) 温度計 JIS B 7411規格群で規定する一般用ガラス製棒状温度計の50度温度計。
42
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
d) 溶存酸素計 温度補償回路を組み入れたもの。
e) マグネチックスターラ
注記 溶存酸素計は,隔膜の取付け,電解液の交換,電極の消耗による交換などの方法が,計測器に
よって異なる。特に,隔膜の取付けは,測定に大きな影響を与えるので注意する。取扱説明書
に従って正しく行う。
14.3.3
準備操作
準備操作は,次による。
a) 溶存酸素計に隔膜電極を接続し,約30分間通電しておく。
b) 校正用容器に,試料と同じ温度に調節した14.3.1 a) 1) の亜硫酸ナトリウム溶液を注入し,マグネチッ
クスターラで静かにかき混ぜながら隔膜電極を挿入し,指示値が安定してから指示値をゼロに合わせ
る。ただし,ゼロ校正時には,溶存酸素計の測定レンジが最小に,又は自動レンジ切替え方式では最
小レンジに切り替わっていることを確認してから行う。
注記 指示が安定するまで通常,2〜5分間を要する。
c) 隔膜電極及び温度計を取り出し,水でよくすすぎ,溶存酸素測定容器に挿入する。
なお,b) からc) に移るときには,隔膜電極を特によく洗浄する。
d) 注入管の一端から溶存酸素測定容器の底部に静かに溶存酸素飽和水を注入し,溶存酸素測定容器の容
量の25〜50 %を流出させた後,排出管の先端を閉じる。溶存酸素飽和水は,溶存酸素測定容器内で調
製してもよい。
e) マグネチックスターラでかき混ぜながら,溶存酸素計の指示値が安定するのを待つ。かき混ぜ速度に
よって指示値に差が生じるので,できるだけスパン調節操作時と同じ条件に保つ。スパン校正時には,
溶存酸素計の測定レンジが最大に,又は自動レンジ切替え方式では最大レンジに切り替わっているこ
とを確認してから行う。
f)
b)〜e) の操作を2,3回繰り返して指示値が,それぞれゼロ及び溶存酸素の飽和量に合致しているこ
とを確かめる。
14.3.4
操作
操作は,次による。
a) 14.3.3 d) に準じて,試料を溶存酸素測定容器の底部に,気泡が入らないように静かに注入する。
なお,溶存酸素測定瓶又は培養瓶などを測定容器として用いる場合には,サイホンを用いて静かに
試料を測定用器にとり,直ちに電極及び温度計を挿入して測定する。また,温度計を挿入する代わり
に,電極に内装されている温度計で温度を測定してもよい。
b) マグネチックスターラでかき混ぜながら,温度計の目盛を確認し,次に,溶存酸素計の指示値の安定
するのを待って指示値(O:μg/L)を読み取る。
注記 指示値は,温度1 ℃の上昇につき約5 %増大する。
c) かき混ぜ速度によって指示値に差が生じるので,できるだけスパン調節操作時と同じ条件に保つ。
d) 測定中に試料を一定の流量で流す場合には,マグネチックスターラでかき混ぜる必要はない。指示値
は,一般に試料の流速に依存する。製造業者の示した電極面の流速を保つように調節する。
e) 試料の溶存酸素の濃度が低く,また,試料の量が十分に採取できる場合には,ほかの試験方法(例え
ば,14.2など)で確認することが望ましい。
14.3.5
留意事項
a) 隔膜電極法による溶存酸素計には,ガルバニ電池方式とポーラログラフ方式とがある。ガルバニ電池
43
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
方式は,金,銀などの貴金属と,鉛,亜鉛などの卑金属とを組み合わせ,電解質溶液(塩化カリウム
又は水酸化カリウムの溶液)に浸すと,卑金属は溶解し,酸素が還元されて電極間に電流が流れるこ
とによる。酸素がない場合は,電池の分極作用で電流が流れない。いずれの方式も隔膜の酸素拡散係
数,電解質溶液の種類,反応速度などによって測定感度及び再現性が異なる。
b) 溶存酸素O:1 mg/L未満を測定する場合には,溶存酸素の濃度約440 µg/Lの溶液を調製し,ゼロ校正
液及びスパン校正液を用いて3点での直線性を確認する。
c) 酸素の濃度約400 µg/Lの溶液は,水1 Lを20 ℃,101.325 kPaに保って,国家計量標準にトレーサブ
ルな酸素のNO 1を流量約1 L/minで球形又は板状のガラスろ過板G2又はG3を用いて40分間以上通
気し,溶解させる。このときの溶存酸素の濃度はO:443 µg/Lとなる。
d) 気圧101.325 kPaで水温20 ℃の水1 Lに窒素(窒素1級)及び校正ガス(酸素濃度0.05及び0.5 vol%)
を流量約1 L/minでJIS R 3503で規定するガラスろ過板2又は3(球形又は板状)を用いて90分間通
気したときの溶存窒素及び溶存酸素の濃度の変化を測定した一例を,図10に示す。
図10−窒素及び校正ガスを通気したときの溶存酸素の濃度変化の測定例
e) 使用前及び測定時の隔膜電極の取扱いは,隔膜の表面に指を触れない。
f)
電解液及び隔膜を交換した場合,又は隔膜を乾燥させてしまった場合は,隔膜を水で湿し,14.3.3の
準備操作を行う前に指示値が安定するように時間をおく。
g) 試料に浸したときに気泡が電極に付着していないことを確認する。
h) 酸素が常に供給されるように,試料は隔膜上を絶えず流れていることが必要である。また,指示値の
振れが生じない程度の流量であることを確認する。
14.4
光学式センサ法
光学式センサを試料に浸せきして溶存酸素濃度を測定する。光学式センサは,蛍光物質又はりん光物質
経過時間
(min)
校正ガスの種類
窒素1級
酸素0.05 vol %
(バランス窒素)
酸素0.5 vol %
(バランス窒素)
0
5
10
15
20
30
45
60
75
90
9 020
2 820
497
172
93
41
12
4.3
1.4
0.8
9 020
2 880
570
304
136
54
25
20
20
21
9 010
3 000
594
399
283
216
211
210
210
212
通水条件
水温:20 ℃
校正ガス流量:1 L/min
循環水量:0.5 L/min
保有水量:1 L
10000
1000
100
10
1
0.1
0
20
40
60
80
100
校正ガス通気時間(min)
○:窒素1級
△:酸素0.05 vol %
(バランス窒素)
□:酸素0.5 vol %
(バランス窒素)
溶
存
酸
素
濃
度
(μg/L)
44
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
を塗布したセンサキャップ,励起光源,光検出部などから構成され,試料中で励起された蛍光物質又はり
ん光物質が発する光を測定する。試料中に酸素が存在すると消光作用によって発光量が減少するが,この
消光作用は酸素濃度に比例する。光学式センサ法では,発光の位相差,持続時間などから酸素による消光
作用を測定し,溶存酸素濃度を定量する。
定量範囲:1 μg/L以上,繰返し精度:2〜10 %
14.4.1
試薬
試薬は,次による。
a) ゼロ校正液 14.3.1 a) による。
注記 水蒸気飽和させ,JIS K 1107で規定する窒素雰囲気中で,ゼロ調整を行ってもよい。
b) スパン校正液 14.3.1 b) による。
14.4.2
器具及び装置
器具及び装置は,次による。
a) 溶存酸素測定容器 14.3.2 a) による。
b) 校正用容器 14.3.2 b) による。
c) 光学式センサ法溶存酸素計 溶存酸素濃度をμg/L単位で直接表示できるもの,又は酸素の飽和百分率
を%表示できるもの。ほとんどの計器は温度補償,大気圧補償を自動で行い,また,入力した試料の
塩分濃度を濃度計算に反映させる機能をもつ。このような自動機能をもたない計器を使用する場合は,
使用者が温度及び圧力の影響を補正する必要がある。B.5参照。この酸素計の例を図11に示す。
d) 温度計 14.3.2 c) による。ただし,通常,温度センサは溶存酸素計に組み込まれている。
e) 気圧計 1 hPa単位のもの。ただし,通常,気圧計は溶存酸素計に組み込まれている。
図11−光学式センサ法溶存酸素計の構成例
14.4.3
準備操作
準備操作は,次による。
a) ゼロ点の確認 溶存酸素測定容器に14.3.1 a) 1) の亜硫酸ナトリウム溶液を注入し,マグネチックスタ
ーラで静かにかき混ぜながらプローブを挿入し,計器のゼロ点を確認する。ただし,かき混ぜる際に
は,大気から酸素が混入しないように注意し,応答時間が最短化されるようにかき混ぜる。最新のプ
ローブは1〜2分間で安定な指示を得る。製造業者によって応答時間が異なるので,製造業者の取扱説
明書を参照する。
b) スパン側の校正 14.3.1 b) のスパン校正液を使用し,14.3.3のd) 及びe) の校正を実施する。
45
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注記 計器が校正不能となった場合,計器がセンサキャップを受け付けない場合,応答が不安定又
は遅くなった場合は,製造業者の取扱説明書を参照してセンサキャップを交換する。
14.4.4
操作
操作は,次による。
a) 14.4.3 a) に準じて,試料を溶存酸素測定容器の底部に,気泡が入らないように静かに注入する。溶存
酸素測定瓶,培養瓶などを測定容器として用いる場合には,サイホンを用いて静かに試料を容器にと
り,直ちにプローブを挿入して測定する。
b) マグネチックスターラなどを用いて穏やかにかき混ぜ,指示値が安定するのを待って,溶存酸素の濃
度(O:μg/L)又は飽和百分率(%)を読み取る。測定値に影響を与える,試料温度,大気圧などを確
認する。
14.4.5
留意事項
光学式センサ法のスパン側の校正方法について,次に示す。
センサはメンテナンスを実施した後は,必ず校正を実施する。
なお,簡易的な方法として,スパン溶液を使用する代わりにセンサを大気中に開放した状態で,スパン
校正を行うことも推奨されている。この場合は,製造業者の取扱説明書に従って操作を行う。
a) 直接値入力によるセンサの校正 この手順は,サンプルラインを流れている既知濃度の液体サンプル
を活用して校正する。
b) 大気によるセンサの校正 大気スパン校正は大気中の酸素を基準に行う。センサを大気中にさらし,
乾いた状態で校正する。
14.5
溶存酸素プロセス用分析装置による測定方法
溶存酸素プロセス用分析装置(隔膜電極法又は光学式センサ法)によって試料中の溶存酸素を連続的に
測定する。
定量範囲:14.3隔膜電極法,14.4光学式センサ法による。
試験目的によって低濃度,高濃度のレンジ切り替えができるもの(例えば,0〜20 µg/L,0〜200 µg/Lな
ど)を用いるとよい。
14.5.1
試薬
試薬は,次による。
a) ゼロ校正液 隔膜電極法は14.3.1 a),光学式センサ法は14.4.1 a) による。
b) スパン校正液 隔膜電極法は14.3.1 b),光学式センサ法は14.4.1 b) による。
14.5.2
器具及び装置
器具及び装置は,次による。
a) 溶存酸素自動計測器 溶存酸素自動計測器は,検出部,指示部及び記録部で構成する。構成の一例を
図12示す。
図12−溶存酸素プロセス用分析装置の構成の一例
試料採取装置
検出部
指示部
記録部
排出水
46
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
14.5.3
隔膜式電極法による測定
測定は,次による。
14.5.3.1 測定準備
測定準備は,次による。
a) あらかじめ電極を90分間以上水に浸した後,そのままの状態で指示部に接続する。
b) 自動計測器の各部を点検し,特に水漏れのないことを確認してから,所定の手順に従って電源を入れ,
各部が安定するまで30分間以上おく。
14.5.3.2 校正
校正は,次による。この操作は,1〜3か月間に1回程度行うことが望ましい。
a) 自動計測器が安定状態に達した後,電極をゼロ校正液に浸し,指示値が安定した後,ゼロ調節を行う。
または,電気的なゼロ調節を行う。
b) 電極をスパン校正液に浸し,指示値が安定した後,スパン調節を行う。
なお,ゼロ調節に,ゼロ校正液を用いた場合には,電極に付着したゼロ校正液を水で十分にすすい
だ後,電極をスパン校正液に浸す。
14.5.3.3 測定
測定は,次による。
a) 電極を試料で十分にすすいだ後,試料を所定の流量で流す。
b) 指示値が安定した後,連続測定を行う。
c) 試料の溶存酸素の濃度が低く,また,試料の量が十分に採取できる場合には,ほかの試験方法(例え
ば,14.2など)で確認することが望ましい。
14.5.3.4 点検・整備
点検・整備は,定期的に行うこととし,次による。
a) 試料の流量・温度・圧力(1〜3か月適時)
b) 電極の汚れ(1〜3か月適時)
c) 電解液の補給,隔膜の交換(3〜6か月適時)
d) 記録紙及びインクの補給又は交換(適時)
14.5.4
光学式センサ法による測定
測定は,次による。
14.5.4.1 測定準備
測定準備は,次による。
a) 自動計測器の各部を点検し,特に水漏れのないことを確認する。
b) 所定の手順に従って電源を入れる。
14.5.4.2 校正
校正は,次による。この操作は,1年間に1回程度行うことが望ましい。
a) プロセス用分析装置が安定状態に達した後,14.5.1 a) を用いてゼロ点の確認及び14.5.1 b) を用いてス
パン側の校正の操作を行う。
14.5.4.3 測定
測定は,次による。
a) 電極を試料で十分にすすいだ後,試料を所定の流量で流す。
b) 指示値が安定した後,連続測定を行う。
47
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
14.5.4.4 点検・整備
点検・整備は,定期的に行うこととし,次による。
a) 試料の流量・温度・圧力(1〜3か月適時)
b) 電極の汚れ(1〜3か月適時)
15
塩化物イオン(Cl−)
15.1
一般事項
塩化物イオンの定量には,チオシアン酸水銀(II)吸光光度法,塩化銀比濁法,硝酸銀滴定法,イオン
電極法,イオンクロマトグラフ法又は流れ分析法を適用する。
15.2
チオシアン酸水銀(II)吸光光度法
試料にチオシアン酸水銀(II)と硫酸アンモニウム鉄(III)を加えると,チオシアン酸水銀(II)のチオ
シアン酸イオンと塩化物イオンとが置換され,チオシアン酸イオンが遊離する。このチオシアン酸イオン
と鉄(III)とが反応して生じる赤だいだいの錯体の吸光度を測定して,塩化物イオンを定量する。
定量範囲: 10 mmセルの場合Cl−:0.4〜10 mg/L
20 mmセルの場合Cl−:0.2〜5 mg/L
50 mmセルの場合Cl−:0.1〜2 mg/L
100 mmセルの場合Cl−:0.05〜1 mg/L
繰返し精度:2〜10 %
15.2.1
試薬
試薬は,次による。
a) 水 JIS K 0557で規定するA3又はA4の水。
b) 硫酸アンモニウム鉄(III)溶液 JIS K 8982で規定する硫酸アンモニウム鉄(III)・12水(鉄みょう
ばん)60 gを硝酸(5 mol/L)(JIS K 8541で規定する硝酸380 mLに水600 mLを加え,室温まで冷却
し,更に水を加えて1 Lとする。)1 Lに溶かす。濁りがあればろ過し,着色瓶に入れて保存する。
c) チオシアン酸水銀(II)エタノール溶液 チオシアン酸水銀(II)1.5 gをJIS K 8102で規定するエタ
ノール(95)500 mLに溶かし,着色ガラス瓶に入れて保存する。
d) 塩化物イオン標準液(Cl−:1 000 mg/L) 国家計量標準にトレーサブルな塩化物イオン標準液(Cl−
1 000 mg/L)を使用するか,又は,次による。JIS K 8005で規定する容量分析用標準物質の塩化ナト
リウムをあらかじめ600 ℃で約1時間加熱し,デシケータ中で放冷する。NaCl 100 %に対してその
1.648 gをとり,少量の水に溶かし,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
e) 塩化物イオン標準液(Cl−:10 mg/L) 塩化物イオン標準液(Cl−:1 000 mg/L)10 mLを全量フラス
コ1 000 mLにとり,水を標線まで加える。
15.2.2
器具及び装置
器具及び装置は,次による。
a) ガラス器具類 4.9 a) で規定するガラス器具を使用前に水で洗う。
b) メスシリンダ(有栓形) JIS R 3505で規定するもの。
c) 光度計 分光光度計又は光電光度計。
15.2.3
操作
操作は,次による。ただし,吸収セル100 mmを用いる場合には,試料100 mLをとり,これに使用する
試薬は,それぞれ2倍量を用いる。
48
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) 5.3によってろ過した試料50 mLをメスシリンダ(有栓形)にとる。塩化物イオン濃度が10 mg/L以
上の場合は,試料の適量をとり,水を加えて50 mLとする。
b) 硫酸アンモニウム鉄(III)溶液10 mLとチオシアン酸水銀(II)エタノール溶液5 mLとを加え,栓を
してよく振り混ぜる。
c) 発色速度は温度によって異なるので,液温を約20 ℃に保ち約10分間放置する。
d) 空試験として,水50 mLについて,b) 及びc) の操作を行う。
e) c) の溶液を吸収セルに移し,d) の空試験の溶液を対照液とし,波長460 nm付近の吸光度を測定する。
f)
あらかじめ,次によって作成した塩化物イオンの検量線から,試料中の塩化物イオンの濃度(Cl−:
mg/L)を算出する。a) で試料の適量をとり,水で50 mLにした場合は,希釈率を考慮して濃度を算
出する。
1) セル長が10 mmの場合,塩化物イオン標準液(Cl−:10 mg/L)2〜50 mL,セル長が20 mmの場合,
塩化物イオン標準液(Cl−:10 mg/L)1〜25 mL,セル長が50 mmの場合,塩化物イオン標準液(Cl −:
10 mg/L)0.5〜10 mLをメスシリンダ(有栓形)50 mLに段階的にとり,水を加えて50 mLとする。
セル長が100 mmの場合,塩化物イオン標準液(Cl−:10 mg/L)0.5〜10 mLをメスシリンダ(有栓
形)100 mLに段階的にとり,水を加えて100 mLとする。b)〜e) の操作を行って塩化物イオン濃度
(Cl−:mg/L)と吸光度との関係線を作成する。
なお,検量線作成時は,試料と標準液との温度差が±2 ℃になるようにする。
15.2.4
留意事項
a) 臭化物イオン,よう化物イオン,シアン化物イオンなどは妨害する。チオ硫酸イオン,硫化物イオン
及び亜硫酸イオンも妨害するので,あらかじめ酸化しておく。
b) 塩化物イオンは広く存在するので,手の汗などからの汚染及び実験室の空気などからの汚染に注意す
る。
警告 水銀化合物を使用するため,法令に従い安全及び健康に対する適切な措置を取らなければな
らない。また,廃液の処分には特に注意する。
15.3
塩化銀比濁法
試料に硝酸を加えて硝酸酸性とした後,硝酸銀溶液を加え塩化物イオンを塩化銀コロイドとし,コロイ
ドの散乱によって減少する透過光の強度を見かけの吸光度として測定し,塩化物イオン濃度を定量する。
定量範囲: 10 mmセルの場合Cl−:0.5〜10 mg/L
50 mmセルの場合Cl−:0.1〜2 mg/L
繰返し精度:10 %以下
15.3.1
試薬
試薬は,次による。
a) 水 15.2.1 a) による。
b) 硝酸(2 mol/L) JIS K 8541で規定する質量分率60 %の硝酸15 mLを水に溶解して,全量フラスコ
100 mLに移し入れ,水を標線まで加える。
c) 硝酸銀溶液(0.02 mol/L) JIS K 8550で規定する硝酸銀0.34 gを水に溶解して,全量フラスコ100 mL
に移し入れ,水を標線まで加える。
d) 塩化物イオン標準液(Cl−:1 000 mg/L) 15.2.1 d) による。
e) 塩化物イオン標準液(Cl−:10 mg/L) 15.2.1 e) による。
f)
塩化物イオン標準液(Cl−:5 mg/L) 塩化物イオン標準液(Cl−:10 mg/L)50 mLを全量フラスコ
49
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
100 mLにとり,水を標線まで加える。
15.3.2
器具及び装置
器具及び装置は,次による。
a) 光度計 分光光度計又は光電光度計。
b) メスシリンダ(有栓形) 15.2.2 b) による。
15.3.3
操作
操作は,次による。
a) 試料を1 μmガラス繊維ろ紙でろ過し,初めのろ液約50 mLを捨て,次のろ液を採取する。ろ過した
試料25 mlをメスシリンダ(有栓形)50 mLにとる。塩化物イオン濃度が10 mg/L以上の場合は,試
料の適量をとり,水を加えて25 mLとする。
b) 硝酸(2 mol/L)1 mLを加えてメスシリンダ(有栓形)をよく振り混ぜ,硝酸銀溶液(0.02 mol/L)5 mL
を加えて再びよく振り混ぜる。
c) 液温を15〜35 ℃に保ち10分間放置する。呈色は,試薬溶液添加後,約30分間は安定である。
d) 空試験として,水25 mLについて,b) 及びc) の操作を行う。
e) c) の溶液を吸収セルに移し,d) の空試験の溶液を対照液とし,波長440 nm付近の透過光の強度を見
かけの吸光度として測定する。
f)
あらかじめ,次によって作成した塩化物イオンの検量線から,試料中の塩化物イオンの濃度(Cl−:
mg/L)を算出する。a) で試料の適量をとり,水で25 mLにした場合は,希釈率を考慮して濃度を算
出する。
1) セル長が10 mmの場合,塩化物イオン標準液(Cl−:10 mg/L)1.25〜25 mL,セル長が50 mmの場
合,塩化物イオン標準液(Cl−:5 mg/L)0.5〜10 mLをメスシリンダ(有栓形)50 mLに段階的にと
り,水を加えて25 mLとした後,b)〜e) の操作を行って塩化物イオン濃度(Cl−:mg/L)と吸光度
との関係線を作成する。
15.3.4
留意事項
a) 臭化物イオン,よう化物イオン,シアン化物イオン,チオ硫酸イオン及び硫化物イオンは妨害する。
亜硫酸イオンは妨害するが,あらかじめ過酸化水素(1+1)(JIS K 8230で規定する過酸化水素を用い
て調製する。)で酸化すれば妨害しない。
b) 塩化物イオンは広く存在するので,手の汗などからの汚染及び実験室の空気などからの汚染に注意す
る。
c) 15 ℃以下の低温又は40 ℃以上の高温で負の誤差を生じる場合があるので液温の管理に注意する。
d) ポリアクリル酸,ポリマレイン酸,グルコン酸などのカルボン酸系分散剤が含まれる場合は,妨害す
る場合がある。この場合は標準添加法によって分析する。
e) 着色した試料については,妨害する場合があるので注意する。鉄による着色は,15.3.3 a) のろ過をす
れば妨害しない。有機物による着色で妨害する場合には,他の分析方法を採用する。
f)
試料のpHは,通常のボイラの給水及びボイラ水である中性付近からpH 12の間であれば測定に影響
しない。
g) ガラス繊維ろ紙以外のろ過材としては,孔径1 μm以下のセルロースアセテート又はPTFEメンブレン
フィルタが使用できる。ろ紙5種Cは負の妨害をするため使用しない。
15.4
硝酸銀滴定法
試料のpHを約7に調節し,2',7'-ジクロロフルオレセイン二ナトリウム[9-(2-カルボキシフェニル)-2,7-
50
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ジクロロ-6-ヒドロキシ-3H-キサンテン-3-オン二ナトリウム塩]又はウラニン(フルオレセインナトリウム)
溶液を指示薬として,硝酸銀溶液で滴定して塩化物イオンを定量する。
定量範囲:Cl−:100 mg/L以上
15.4.1
試薬
試薬は,次による。
a) 硝酸(1+65) JIS K 8541で規定する硝酸を用いて調製する。
b) 炭酸ナトリウム溶液(50 g/L) JIS K 8625で規定する炭酸ナトリウム5 gを水に溶かして100 mLと
する。
c) ジクロロフルオレセインナトリウム溶液(2 g/L) 2',7'-ジクロロフルオレセイン二ナトリウム0.2 g
を水に溶かして100 mLとする。ジクロロフルオレセインナトリウム溶液(2 g/L)に代えて,JIS K 8830
で規定するウラニン(フルオレセインナトリウム)[9-(2-カルボキシフェニル)-6-ヒドロキシ-3H-
キサンテン-3-オン二ナトリウム]を用い,0.2 gを水100 mLに溶かしたものを用いてもよい。
d) デキストリン溶液 JIS K 8646で規定するデキストリン水和物2 gを水に溶かして100 mLとする。使
用時に調製する。
e) 40 mmol/L硝酸銀溶液 JIS K 8550で規定する硝酸銀6.8 gを水に溶かして1 Lとし,着色ガラス瓶に
保存する。この溶液は使用時に,次によって標定して用いる。
1) JIS K 8005で規定する容量分析用標準物質の塩化ナトリウムをあらかじめ600 ℃で約1時間加熱
し,デシケータ中で放冷する。NaCl 100 %に対してその0.47 gを1 mgの桁まではかりとり,少量の
水に溶かし,全量フラスコ200 mLに移し入れ,水を標線まで加える。
2) この20 mLをビーカにとり,水を加えて液量を約50 mLとし,これにデキストリン溶液5 mLとジ
クロロフルオレセインナトリウム溶液(2 g/L)1,2滴を加え,静かにかき混ぜながらこの硝酸銀溶
液で滴定する。黄緑の蛍光が消失して僅かに赤くなったときを終点とする。次の式によって40
mmol/L硝酸銀溶液のファクタ( f )を算出する。
7
337
002
.0
1
200
20
100
×
×
×
×
=
x
b
a
f
ここに,
a: 塩化ナトリウムの量(g)
b: 塩化ナトリウムの純度(%)
x: 滴定に要した40 mmol/L硝酸銀溶液(mL)
0.002 337 7: 40 mmol/L硝酸銀溶液1 mLの塩化物イオン相当量(g)
15.4.2
操作
操作は,次による。
a) 5.3によってろ過した試料50 mL(Cl−:20 mg以上を含む場合には適量をとり,水を加えて50 mLと
する。)をビーカ又は磁器皿にとる。ただし,試料に著しい濁りが認められない場合は,ろ過操作を省
略してもよい。
b) 試料が酸性の場合には,炭酸ナトリウム溶液(50 g/L)で,また,アルカリ性の場合には硝酸(1+65)
を用いてpHを約7に調節する。
c) デキストリン溶液5 mL及びジクロロフルオレセインナトリウム溶液(2 g/L)1,2滴を加えてかき混
ぜる。
d) 静かにかき混ぜながら40 mmol/L硝酸銀溶液で滴定する。黄緑の蛍光が消失して僅かに赤くなったと
きを終点とする。
51
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
e) 次の式によって試料中の塩化物イオンの濃度(Cl−:mg/L)を算出する。
418
.1
000
1
×
×
×
=
V
f
a
C
ここに,
C: 塩化物イオン(Cl−:mg/L)
a: 滴定に要した40 mmol/L硝酸銀溶液(mL)
f: 40 mmol/L硝酸銀溶液のファクタ
V: 試料(mL)
1.418: 40 mmol/L硝酸銀溶液1 mLの塩化物イオン相当量(mg)
15.4.3
留意事項
a) 臭化物イオン,よう化物イオン,シアン化物イオンなどが共存すると,塩化物イオンとして定量され
る。亜硫酸イオン,チオ硫酸イオン,硫化物イオンも妨害するが,あらかじめ過酸化水素で酸化すれ
ば妨害しない。
b) 塩化物イオン濃度が低い場合は,試料50 mL中に塩化物イオンを5 mg以上含むように,15.2.1 d) の
塩化物イオン標準液(Cl−:1 000 mg/L)を試料に加え,15.4.2のb)〜e) の操作を行う。この場合は,
試料と同量の塩化物イオン標準液を加えた水50 mLについても同様に15.4.2のb)〜e) の操作を行っ
て,試料の場合の滴定値を補正する。
15.5
イオン電極法
試料に酢酸塩緩衝液を加えてpHを約5に調節し,塩化物イオン電極を指示電極として電位を測定し,
塩化物イオンを定量する。
定量範囲:Cl−:5〜1 000 mg/L,繰返し精度:5〜20 %
15.5.1
試薬
試薬は,次による。
a) 水 15.2.1 a) による。
b) 酢酸塩緩衝液(pH 5) JIS K 8548で規定する硝酸カリウム100 gとJIS K 8355で規定する酢酸50 mL
とを水500 mLに加えて溶かし,これに水酸化ナトリウム溶液(100 g/L)を加え,pH計を用いてpH
を5に調節し,水を加えて1 Lとする。
c) 塩化物イオン標準液(Cl−:1 000 mg/L) 15.2.1 d) による。
d) 塩化物イオン標準液(Cl−:100 mg/L) 塩化物イオン標準液(Cl−:1 000 mg/L)20 mLを全量フラ
スコ200 mLにとり水を標線まで加える。
e) 塩化物イオン標準液(Cl−:10 mg/L) 塩化物イオン標準液(Cl−:100 mg/L)20 mLを全量フラスコ
200 mLにとり,水を標線まで加える。使用時に調製する。
f)
塩化物イオン標準液(Cl−:5 mg/L) 塩化物イオン標準液(Cl−:100 mg/L)10 mLを全量フラスコ
200 mLにとり,水を標線まで加える。使用時に調製する。
15.5.2
器具及び装置
器具及び装置は,次による。
a) 電位差計 0.1 mV又はそれ以下の電位差を読み取れるもの。高入力抵抗電位差計(例えば,デジタル
式pH-mV計,拡大スパン付pH-mV計,イオン電極用電位差計など)。
b) イオン濃度計
c) 指示電極 塩化物イオン電極。標準液を用いた起電力の応答は,25 ℃における塩化物イオン濃度の
10倍濃度変化当たり55 mV以上,液温10〜30 ℃で塩化物イオン濃度5 mg/L以上での応答時間が1
分間以内のもの。塩化物イオン電極の感応膜が汚れたり,きずついたりすると,指示値の応答速度が
52
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
遅くなったりするので,製造業者の取扱説明書に基づき電極を保持する。また,使用に際しては,塩
化物イオン標準液(Cl−:5 mg/L)に浸し,指示値が安定しているかを確認する。
d) 参照電極 銀-塩化銀電極を用いる。二重液絡形のもので,外筒液には硝酸カリウム溶液(100 g/L)(JIS
K 8548で規定する硝酸カリウムを用いて調製する。)を用いるか,又は,イオン電極用セラミック形
を用いる。
e) 測定容器 試料100 mLで扱えるもの。恒温ジャケットを取り付けたもの。
f)
恒温槽 測定容器のジャケットに水温25±0.2 ℃の水を供給できるもの。
g) マグネチックスターラ 回転による発熱で液温に変化を与えないもの。
15.5.3
操作
15.5.3.1 塩化物イオン電極を用いる場合
操作は,次による。
a) 試料100 mLを測定容器にとり,試料のpHとイオン強度調節のために酢酸塩緩衝液(pH 5)10 mLを
加える。ただし,試料が酸性の場合には,水酸化ナトリウム溶液(40 g/L),アルカリ性の場合には,
酢酸(1+10)で,あらかじめpHを約5に調節する。また,試料に硫化物イオンが含まれている場合
には,あらかじめ,酢酸亜鉛溶液(100 g/L)を加えて,硫化物イオンを固定してろ紙でろ過し,ろ液
のpHを約5に調節する。
b) 恒温槽から水を送り,測定容器の液温を25±0.5 ℃に調節する。
c) 指示電極と参照電極とを浸し,マグネチックスターラで泡が電極に触れない程度に強くかき混ぜる。
d) 液温をはかり,電位差計で電位を測定する。
e) あらかじめ,次によって作成した塩化物イオンの検量線から,試料中の塩化物イオンの濃度(Cl−:
mg/L)を算出する。
1) 塩化物イオン標準液(Cl−:5 mg/L)100 mLを測定容器200 mLにとり,酢酸塩緩衝液(pH 5)10 mL
を加える。
2) b)〜d) の操作を行い,電位を測定する。
3) 塩化物イオン標準液(Cl−:10 mg/L)100 mL,塩化物イオン標準液(Cl−:100 mg/L)100 mL及び
塩化物イオン標準液(Cl−:1 000 mg/L)100 mLをそれぞれ測定容器にとり,これに酢酸塩緩衝液
(pH 5)10 mLを加える。b) 及びd) の操作を行ってそれぞれの塩化物イオン標準液の電位を測定す
る。
4) 横軸に塩化物イオンの濃度の対数を,縦軸に電位をとり,塩化物イオンの濃度(Cl−:mg/L)と電
位との関係線を作成する。塩化物イオン標準液(Cl−:10 mg/L)と塩化物イオン標準液(Cl−:1 000
mg/L)との電位の差は,110〜120 mV(25 ℃)の範囲に入り,塩化物イオンの濃度Cl−:5〜1 000 mg/L
の間の検量線は直線になる。
15.5.3.2 イオン濃度計を用いる場合
操作は,次による。
a) 塩化物イオン標準液(Cl−:10 mg/L),塩化物イオン標準液(Cl−:1 000 mg/L)を別々の測定容器に
とり,それぞれに酢酸塩緩衝液(pH 5)10 mLを加える。
b) 15.5.3.1のb) 及びc) の操作を行ってイオン濃度計の指示値をCl−:10 mg/LとCl−:1 000 mg/Lにな
るように調節する。
c) 塩化物イオン標準液(Cl−:5 mg/L)と塩化物イオン標準液(Cl−:100 mg/L)とを用いてイオン濃度
計の指示値を確認する。
53
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
d) 15.5.3.1 a) で処理した試料を測定容器にとり,イオン濃度計の指示値を読み,試料の塩化物イオン濃
度(Cl−:mg/L)を求める。
15.5.4
留意事項
1) この方法では硫化物イオンなどが妨害する。
2) 主な共存物質の許容限度を塩化物イオンに対する化学当量最大比率で,次に示す。
NO3−,SO42−,PO43− :104
F− :102
Br− :10−2
I−,CN−,S2− :10−3
15.6
イオンクロマトグラフ法
試料中の陰イオンをイオンクロマトグラフ法によって定量する。検出器には電気伝導率検出器を用いる。
この方法によって,表8に示す陰イオンが同時定量できる。それぞれの陰イオンの定量範囲及び繰返し精
度の例を,表8に示す。
定量範囲:サプレッサ法の場合Cl−:0.05〜25 mg/L,ノンサプレッサ法の場合Cl−:0.1〜25 mg/L,繰返
し精度:2〜10 %(装置,測定条件によって異なる。)
表8−各陰イオンの定量範囲及び繰返し精度の例a)
対象陰イオン
定量範囲
mg/L
繰返し精度
%
塩化物イオン(Cl−)b)
0.1〜25
2〜10
硫酸イオン(SO42−)b)
1〜100
2〜10
注a) 定量範囲は,検出器,試料注入量,カラムのイオン交換容量などによっ
て変わる。
b) サプレッサと組み合わせる方式の場合には,Cl−:0.05〜25 mg/L,
SO42−:0.2〜100 mg/L
15.6.1
試薬
試薬は,次による。
a) 水 15.2.1 a) による。
b) 溶離液 溶離液は,装置の種類及び分離カラムに充塡した陰イオン交換体の種類によって異なるので,
あらかじめ塩化物イオン,亜硝酸イオン,臭化物イオン,硝酸イオン及び硫酸イオンの分離を15.6.3
の準備操作を行って確認する。
c) 再生液 再生液は,サプレッサ法の場合に使用するが,装置の種類及びサプレッサの種類によって再
生液が異なる。あらかじめ分離カラムと組み合わせて15.6.3の準備操作を行って再生液の性能を確認
する。
d) 塩化物イオン標準液(Cl−:1 000 mg/L) 15.2.1 d) による。
e) 塩化物イオン標準液(Cl−:100 mg/L) 塩化物イオン標準液(Cl−:1 000 mg/L)10 mLを全量フラ
スコ100 mLにとり,水を標線まで加える。
f)
亜硝酸イオン標準液(NO2−:1 000 mg/L) 国家計量標準にトレーサブルな亜硝酸イオン標準液(NO2 −
1 000 mg/L)を使用するか,又は,次による。JIS K 8019で規定する亜硝酸ナトリウムを105〜110 ℃
で約4時間加熱し,デシケータ中で放冷した後,亜硝酸ナトリウムの純度をJIS K 8019によって求め
54
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
る。NaNO2 100 %に対して1.500 gに相当する亜硝酸ナトリウムをとり,少量の水に溶かして,全量フ
ラスコ1 000 mLに移し入れ,水を標線まで加える。使用時に調製する。
g) 硝酸イオン標準液(NO3−:1 000 mg/L) 国家計量標準にトレーサブルな硝酸イオン標準液(NO3−1 000
mg/L)を使用するか,又は,次による。JIS K 8548で規定する硝酸カリウムをあらかじめ105±2 ℃
で約2時間加熱し,デシケータ中で放冷する。その1.631 gをとり,少量の水に溶かして全量フラスコ
1 000 mLに移し入れ,水を標線まで加える。0〜10 ℃の暗所に保存する。
h) 硫酸イオン標準液(SO42−:1 000 mg/L) 国家計量標準にトレーサブルな硫酸イオン標準液(SO42−
1 000 mg/L)を使用するか,又は,次による。JIS K 8962で規定する硫酸カリウムを約700 ℃で約30
分間加熱し,デシケータ中で放冷する。その1.815 gをとり,少量の水に溶かして全量フラスコ1 000 mL
に移し入れ,水を標線まで加える。使用時に調製する。
i)
陰イオン混合標準液(Cl−:20 mg/L,NO2−:100 mg/L,NO3−:100 mg/L,SO42−:100 mg/L) d) の
塩化物イオン標準液(Cl−:1 000 mg/L)2 mL,f) の亜硝酸イオン標準液(NO2−:1 000 mg/L)10 mL,
g) の硝酸イオン標準液(NO3−:1 000 mg/L)10 mL及びh) の硫酸イオン標準液(SO42−:1 000 mg/L)
10 mLをそれぞれ全量フラスコ100 mLにとり,水を標線まで加える。使用時に調製する。
j)
陰イオン混合標準液(Cl−:10 mg/L,NO2−:10 mg/L,Br−:10 mg/L,NO3−:10 mg/L,SO42−:10 mg/L)
d) の塩化物イオン標準液(Cl−:1 000 mg/L)10 mL,f) の亜硝酸イオン標準液(NO2−:1 000 mg/L)
10 mL,g) の硝酸イオン標準液(NO3−:1 000 mg/L)10 mL及びh) の硫酸イオン標準液(SO42−:1 000
mg/L)10 mL及び臭化物イオン標準液(Br−:1 000 mg/L)[JIS K 8506で規定する臭化カリウムを
105 ℃で約4時間加熱し,デシケータ中で放冷する。その1.489 g(臭素として1.00 g)をとり,少量
の水に溶かし,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。]10 mLをそれぞれ全量フ
ラスコ1 000 mLにとり,水を標線まで加える。使用時に調製する。
15.6.2
器具及び装置
器具及び装置は,次による。
a) イオンクロマトグラフ イオンクロマトグラフには,分離カラムとサプレッサとを組み合わせた方式
のものと,分離カラム単独の方式のものとがある。いずれでもよいが,次に掲げる条件を満たすもの
で,塩化物イオン,亜硝酸イオン,臭化物イオン,硝酸イオン,硫酸イオンなどが分離定量できるも
の。
1) 分離カラム ステンレス鋼製又は四ふっ化エチレン樹脂製,ポリエーテルエーテルケトン製などの
合成樹脂製のものに,強塩基性陰イオン交換体(表層被覆形,全多孔性シリカ形など)を充塡した
もの。
2) サプレッサ 溶離液中の陽イオンの濃度に対して十分なイオン交換能力をもつ陽イオン交換膜(膜
形,電気透析形がある。)又は同様な性能をもった陽イオン交換体(充塡形,サスペンション樹脂吸
着形がある。)を充塡したもの。再生液(再生液が不要のものもある。)と組み合わせて用いる。た
だし,電気透析形の場合は,再生液として検出器からの流出液(検出器から排出される溶液)を用
いる。
3) 検出器 電気伝導率検出器。
4) データ処理部 JIS K 0127の5.7(データ処理部)による。
15.6.3
準備操作
準備操作は,次による。
a) 試料を孔径0.45 µmのろ過膜又はろ紙5種C(又はろ紙6種)でろ過し,初めのろ液約50 mLを捨て,
55
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
その後のろ液をとる。
b) 試料の電気伝導率が10 mS/m(25 ℃)以上の場合には,電気伝導率が10 mS/m以下になるように,
水で一定の割合に薄める。
c) 分離カラムの性能確認を定期的に次の操作によって行う。
溶離液を一定の流量(例えば,1〜2 mL/min)で流し,15.6.1 j) の陰イオン混合標準液(Cl−:10 mg/L,
NO2−:10 mg/L,Br−:10 mg/L,NO3−:10 mg/L,SO42−:10 mg/L)の一定量をイオンクロマトグラ
フに注入し,クロマトグラムを求め,それぞれの陰イオンが分離(分離度1.3程度)できるものを用
いる。分離度(R)は,次の式によって算出する。
(
)
2
1
1
R
2
R
2
W
W
t
t
R
+
−
×
=
ここに,
tR1: 第1ピークの保持時間(s)
tR2: 第2ピークの保持時間(s)
W1: 第1ピークのピーク幅(s)
W2: 第2ピークのピーク幅(s)
試料中の酸性物質,還元性物質の共存などで分離カラムの性能が低下した場合,溶離液の濃度の約
10倍の濃度の溶離液を調製し,分離カラムに注入し洗浄した後,上記の操作を行って性能を確認する。
性能が回復しない場合には,新品と取り替える。
15.6.4
操作
操作は,次による。
a) イオンクロマトグラフを定量できる状態にし,分離カラムに溶離液を一定の流量(例えば,1〜2
mL/min)で流しておく。サプレッサを必要とする装置では再生液を一定の流量で流しておく。
b) 15.6.3のa) 及びb) の準備操作を行った試料の一定量(例えば,50〜200 µLの一定量)をイオンクロ
マトグラフに注入してクロマトグラムを記録する。
c) クロマトグラム上の塩化物イオンに相当するピークについて,ピーク高さ又はピーク面積(指示値)
を読み取る。
d) 試料を薄めた場合には,空試験として試料と同量の水についてa)〜c) の操作を行って試料について得
た指示値を補正する。
e) あらかじめ,次によって作成した塩化物イオンの検量線から,試料中の塩化物イオンの濃度(Cl−:
mg/L)を算出する。
1) 塩化物イオン標準液(Cl−:100 mg/L)0.1〜25 mLを段階的に全量フラスコ100 mLにとり,水を標
線まで加える。この溶液についてa)〜c) の操作を行ってそれぞれの塩化物イオンに相当するピーク
について,指示値を読み取る。別に,空試験として水についてa)〜c) の操作を行ってそれぞれの塩
化物イオンに相当する指示値を補正した後,塩化物イオン濃度(Cl−:mg/L)と指示値との関係線
を作成する。検量線の作成は,試料の測定時に行う。
なお,塩化物イオン以外の陰イオンを同時に試験する場合には,15.6.1 i) の陰イオン混合標準液
(Cl−:20 mg/L,NO2−:100 mg/L,NO3−:100 mg/L,SO42−:100 mg/L)を用いる。
15.6.5
留意事項
a) 塩化物イオンの濃度がCl−:1 mg/Lのとき亜硝酸イオンはNO2−:200 mg/L以下ならば妨害しない。
b) 試料の塩化物イオンの濃度がCl−:0.05 mg/L以下の場合には,濃縮カラム(ステンレス鋼製又は合成
樹脂製のカラム用管に,陰イオン交換体を充塡したもの)で濃縮した後,15.6.4のa)〜e) の操作を行
56
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
い定量する。塩化物イオン濃度がCl−:0.001 mg/L以上の試料を試験する場合には,操作で濃縮カラ
ムに注入する試料の量を40 mL以下の一定量とする。
15.7
流れ分析法
試料中の塩化物イオンを15.2と同様な原理で発色させる流れ分析法によって定量する。また,懸濁物の
多い試料には適用しない。
定量範囲:Cl−:0.1〜15 mg/L,繰返し精度:10 %以下(装置,測定条件によって異なる。)
15.7.1
チオシアン酸水銀(II)発色FIA法
15.7.1.1 試薬
試薬は,次による。調製した溶液は,必要に応じて脱気を行う。
a) 水 15.2.1 a) による。
b) 塩化物イオン発色原液 チオシアン酸水銀(II)0.62 gを,JIS K 8891で規定するメタノール150 mL
に溶解し,水100 mLとJIS K 8541で規定する質量分率60 %の硝酸4 mLとを混合する。さらに,硝
酸鉄(III)・九水和物31 gを水500 mLに溶解したものを混合し,水を加え1 000 mLとする。着色ガ
ラス瓶に入れて保存する。
c) 塩化物イオン発色溶液 塩化物イオン発色原液を水で2倍希釈し,塩化物イオン発色溶液とする。着
色ガラス瓶に入れて保存する。
d) キャリヤ液 水を用いる。
e) 塩化物イオン標準液(Cl−:1 000 mg/L) 15.2.1 d) による。
f)
塩化物イオン標準液(Cl−:100 mg/L) 15.6.1 e) による。
15.7.1.2 器具及び装置
装置の基本構成は,次による(図13参照)。
a) 送液部 脈動の小さいポンプを用いる。
b) 試料導入部 通常6方切替えバルブを用いる。試料注入量は,適切な量を選択する。必要に応じて自
動試料注入装置を用いることができる。
c) 反応部 内径0.5〜1.0 mmの四ふっ化エチレン樹脂などふっ素樹脂製の管及び化学的に不活性でデッ
トボリュームのできるだけ小さな合成樹脂製のジョイントを用いて構成する。
d) 検出部 波長480 nm付近での測定が可能な吸光光度検出器を用いる。
e) 記録部 検出器からの信号を記録できるものを用いる。
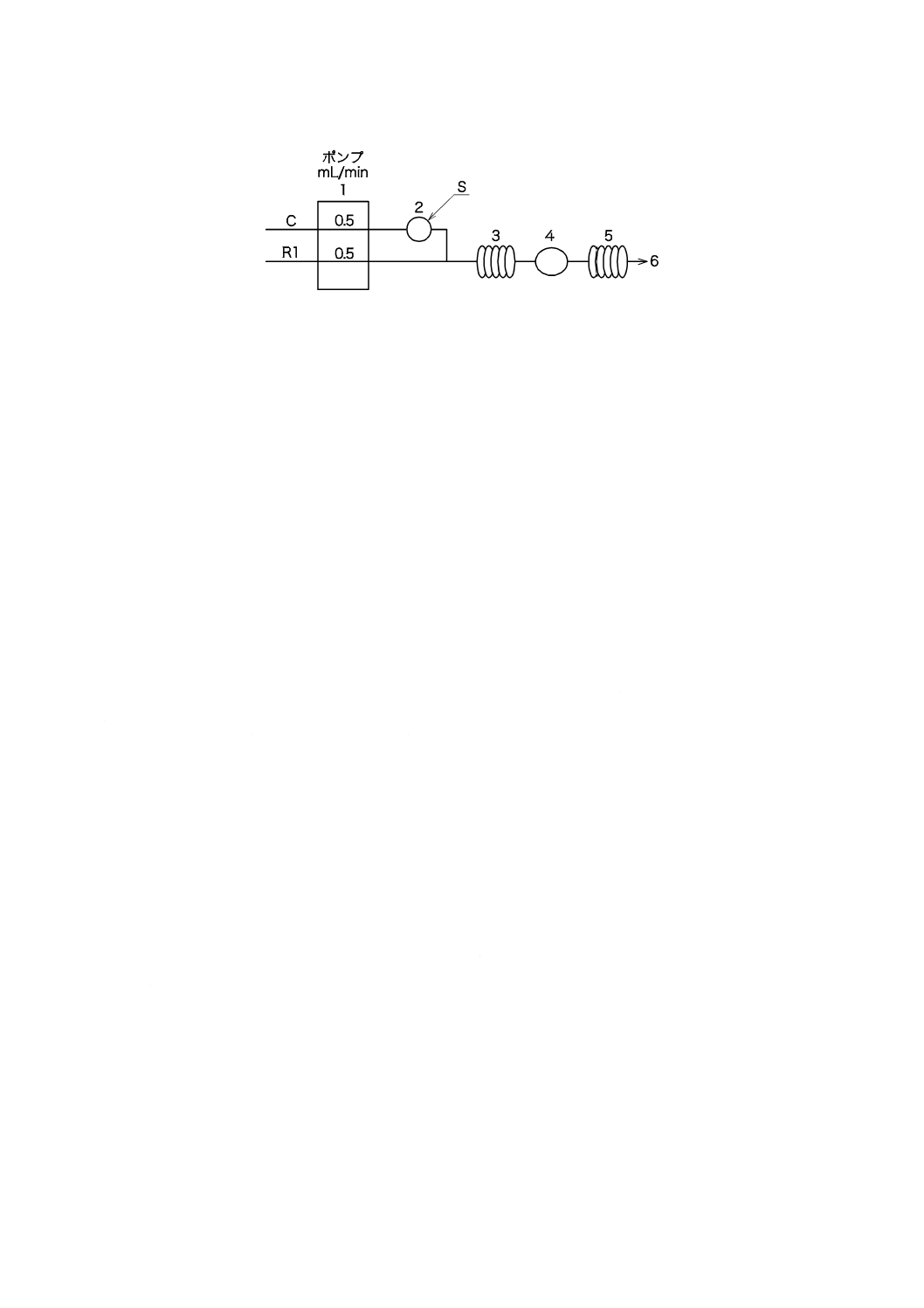
57
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
C: キャリヤ液
R1: 塩化物イオン発色溶液
S: 試料
1: ポンプ
2: 試料導入器(200 μL)
3: 反応コイル(内径0.5 mm 長さ1 m)
4: 検出器(波長480 nm)
5: 背圧コイル(内径0.5 mm 長さ1 m)
6: 廃液
図13−チオシアン酸水銀(II)発色FIA法のシステム例
15.7.1.3 準備操作
準備操作は,次による。
a) 分析装置及び検出器を作動できる状態にし,キャリヤ液及び発色溶液をポンプで送液し,ベースライ
ンが安定するのを待つ。ベースラインのドリフトなどが測定の結果に支障を与えないこと,及び十分
なS/N比が得られることを確認する。
b) 試料の塩化物イオン濃度が検出可能となるように,検出器の感度を調節する。
c) 試料をろ紙5種C,メンブランフィルタ,ガラスフィルタなどでろ過する。
15.7.1.4 操作
操作は,次による。
a) ベースラインの安定,感度などを確認する。
b) 15.7.1.3 c) でろ過した試料の一定量(200 μL)をマイクロシリンジで試料導入器を通してキャリヤ液
に注入する。
c) 検出器による指示値(吸光度又はそれに相当するシグナル)を記録部で記録する。
d) あらかじめ,次によって作成した塩化物イオンの検量線から,試料中の塩化物イオンの濃度(Cl−:
mg/L)を求める。
1) 試料の塩化物イオン濃度を含む濃度範囲の検量線用塩化物イオン標準液の4〜6種類を,15.7.1.1の
e) 又はf) を水で希釈して調製する。
2) 試料の測定時と同じ量の検量線用塩化物イオン標準液を注入して,指示値と塩化物イオン濃度
(Cl −:mg/L)との関係線を作成する。
15.7.1.5 留意事項
用いる機種によっては,発色溶液及びフローダイアグラムが多少異なる場合がある。その場合は,この
規格と機種による場合との相関を確認する。
15.7.2
チオシアン酸水銀(II)発色CFA法
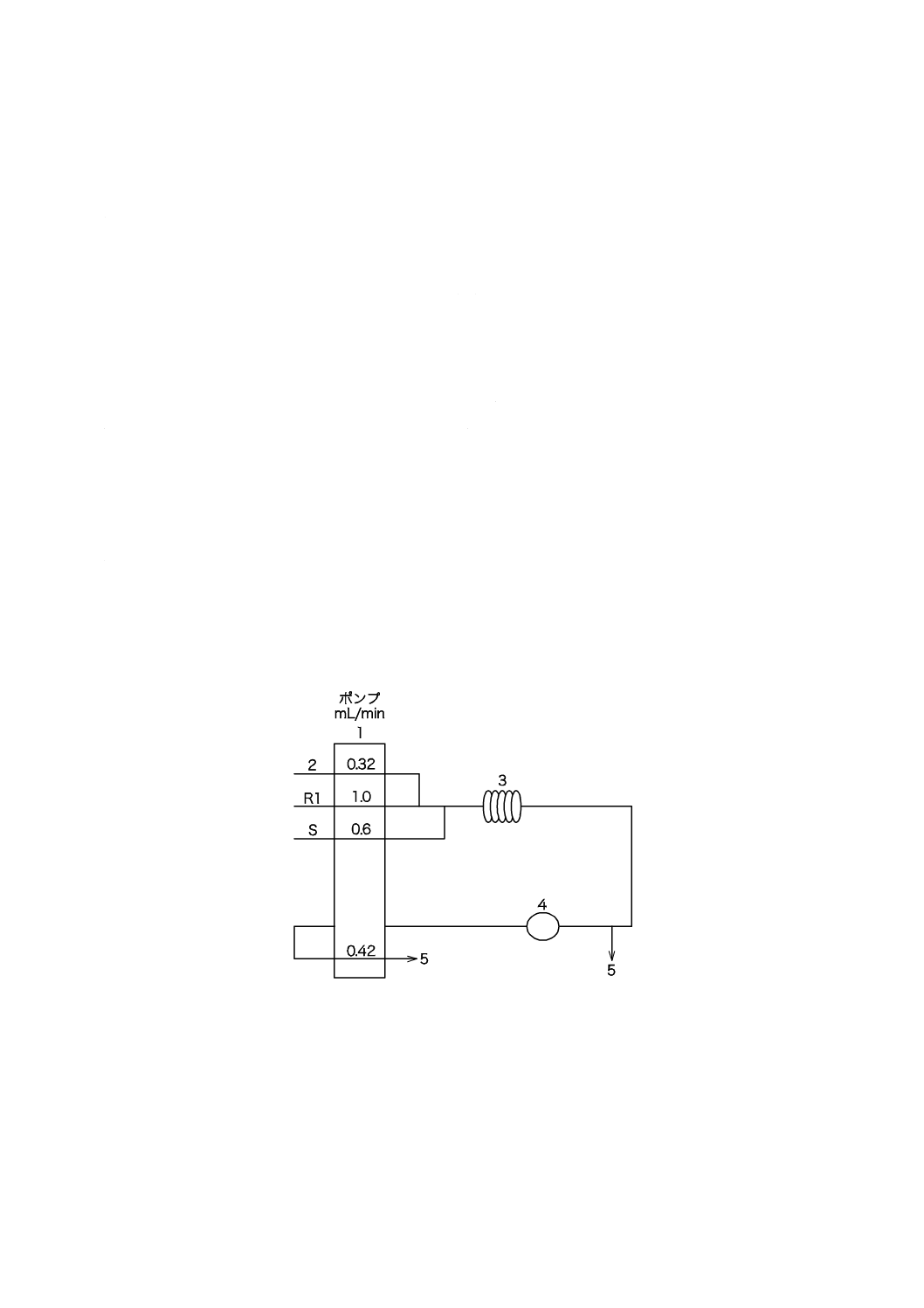
58
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
15.7.2.1 試薬
試薬は,次による。調製した溶液は,必要に応じて脱気を行う。
a) 水 15.2.1 a) による。
b) ポリオキシエチレンドデシルエーテル溶液(300 g/L) ポリオキシエチレンドデシルエーテル30 g
を水に溶かして100 mLとする。
c) 塩化物イオン発色溶液 チオシアン酸水銀(II)0.25 gをJIS K 8891で規定するメタノール150 mLに
完全に溶解する。さらに,水200 mLと硝酸鉄(III)・九水和物10 gとJIS K 8541で規定する硝酸(密
度1.42 g/mL)10 mLを溶解したものを混合し,水を加え1 000 mLとする。これに,ポリオキシエチ
レンドデシルエーテル溶液(300 g/L)2 mLを加えて混合し,塩化物イオン発色溶液とする。着色ガ
ラス瓶に入れて保存する。
d) 塩化物イオン標準液(Cl−:1 000 mg/L) 15.2.1 d) による。
e) 塩化物イオン標準液(Cl−:100 mg/L) 15.6.1 e) による。
15.7.2.2 器具及び装置
装置の基本構成は,次による(図14参照)。
a) 送液部 15.7.1.2 a) による。
b) 試料導入部 再現性がよいものを用いる。
c) 反応部 内径0.5〜2.0 mmの四ふっ化エチレン樹脂製又はガラス製管,及び化学的に不活性でデット
ボリュームのできるだけ小さな合成樹脂製のジョイントを用いて構成する。
d) 検出部 15.7.1.2 d) による。
e) 記録部 15.7.1.2 e) による。
R1: 塩化物イオン発色溶液
S: 試料
1: ポンプ
2: セグメントガス(空気)
3: 反応コイル(内径2 mm 長さ120 cm)
4: 検出器(波長480 nm)
5: 廃液
図14−チオシアン酸水銀(II)発色CFA法のシステム例
59
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
15.7.2.3 準備操作
準備操作は,次による。
a) 分析装置及び検出器を作動できる状態にし,水及び塩化物イオン発色溶液をポンプで送液し,ベース
ラインが安定するのを待つ。空気が一定間隔で流れを分節しているかを確認する。ベースラインのド
リフトなどが測定の結果に支障を与えないこと,及び十分なS/N比が得られることを確認する。
b) 試料の塩化物イオン濃度が検出可能となるように,検出器の感度を調節する。
c) 試料をろ紙5種C,メンブランフィルタ,ガラスフィルタなどでろ過する。
15.7.2.4 操作
操作は,次による。
a) ベースラインの安定,感度などを確認する。
b) 15.7.2.3 c) でろ過した試料を一定流量で試料の流路から流す。
c) 検出器による指示値(吸光度又はそれに相当するシグナル)を記録部で記録する。
d) あらかじめ,次によって作成した塩化物イオンの検量線から,試料中の塩化物イオンの濃度(Cl−:
mg/L)を求める。
1) 試料の塩化物イオン濃度を含む濃度範囲の検量線用塩化物イオン標準液の4〜6種類を,15.7.2.1の
d) 又はe) を水で希釈して調製する。
2) 試料と同様な流量で検量線用塩化物イオン標準液を試料の流路から流して,指示値を記録する。
3) 指示値と塩化物イオン濃度(Cl−:mg/L)との関係線を作成する。
15.7.2.5 留意事項
用いる機種によっては,発色溶液及びフローダイアグラムが多少異なる場合がある。その場合は,本規
格と機種による場合との相関を確認する。
16
亜硫酸イオン(SO32−)
16.1
一般事項
亜硫酸イオンの定量には,よう素滴定法を適用する。
16.2
よう素滴定法
一定量のよう素溶液中に酢酸-酢酸ナトリウム緩衝液を加えた後,試料を加え,次に過剰のよう素をでん
ぷん溶液を指示薬としてチオ硫酸ナトリウム溶液で滴定する。別に,同量の試料をとり,酸性として煮沸
し,亜硫酸イオンを二酸化硫黄として追い出した後,同一の滴定操作を行い,これを空試験値として,チ
オ硫酸イオンなどの還元性物質の影響を補正する。亜硫酸イオンは空気によって酸化されるので,試験は
試料採取直後直ちに行う。
定量範囲:SO32− 2 mg/L以上
16.2.1
試薬
試薬は,次による。
a) 水 50 mmol/Lチオ硫酸ナトリウム溶液,及び10 mmol/Lチオ硫酸ナトリウム溶液を調製する場合は
4.7 b) の溶存酸素を含まない水を用いる。
b) 硫酸(1+35) JIS K 8951で規定する硫酸を用いて調製する。
c) 水酸化ナトリウム溶液(40 g/L) JIS K 8576で規定する水酸化ナトリウム4 gを水に溶かして100 mL
とする。
d) 酢酸-酢酸ナトリウム緩衝液(pH 3.9) JIS K 8371で規定する酢酸ナトリウム三水和物75 gを酢酸(1
60
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
+2)(JIS K 8355で規定する酢酸を用いて調製する。)500 mLに溶かす。
e) エタノール(95) JIS K 8102で規定するもの。
f)
フェノールフタレイン溶液(5 g/L) 9.3.1 a) による。
g) でんぷん溶液(10 g/L) JIS K 8659で規定するでんぷん(溶性)1 gを約水10 mLに混ぜ,熱水100
mL中にかき混ぜながら加え,約1分間煮沸した後,放冷する。使用時に調製する。
h) 窒素 JIS K 1107で規定する2級を用いる。窒素中の酸素の除去は次による。水酸化ナトリウム溶液
(600 g/L)75 mLと水15 mLにJIS K 8780で規定するピロガロール(1,2,3-ベンゼントリオール)5 g
を溶かした溶液とを使用時に混合し,ガス洗浄瓶に入れ,ここに窒素を通気する。
i)
よう素溶液(5 mmol/L) JIS K 8913で規定するよう化カリウム4 gを少量の水に溶かし,これにJIS
K 8920で規定するよう素1.3 gを加えて溶かし,水を加えて1 Lとする。
j)
50 mmol/Lチオ硫酸ナトリウム溶液 JIS K 8637で規定するチオ硫酸ナトリウム五水和物12.5 gとJIS
K 8625で規定する炭酸ナトリウム0.2 gとを4.7 b) の溶存酸素を含まない水に溶かして1 Lとする。2
日間放置した後,標定する。この溶液は使用時に,次によって標定して用いる。
1) JIS K 8005で規定する容量分析用標準物質のよう素酸カリウムを130 ℃で約2時間加熱し,デシケ
ータ中で放冷する。KIO3 100 %に対してその0.357 gをとり,少量の水に溶かし,全量フラスコ200
mLに移し入れ,水を標線まで加える。この20 mLを共栓三角フラスコ300 mLにとり,JIS K 8913
で規定するよう化カリウム2 g及び硫酸(1+5)5 mLを加え,直ちに栓をして静かに振り混ぜ,暗
所に約5分間放置する。これに水100 mLを加え,遊離したよう素をこのチオ硫酸ナトリウム溶液
で滴定する。溶液の黄色が薄くなってから,指示薬としてでんぷん溶液(10 g/L)1 mLを加え,生
じたよう素でんぷんの青い色が消えるまで滴定する。
別に,水について同一条件で空試験を行って補正したmL数から,次の式によって50 mmol/Lチ
オ硫酸ナトリウム溶液のファクタ( f )を算出する。
783
001
.0
1
200
20
100
×
×
×
×
=
x
b
a
f
ここに,
a: よう素酸カリウムの量(g)
b: よう素酸カリウムの純度(%)
x: 滴定に要した50 mmol/Lチオ硫酸ナトリウム溶液(補正
した値)(mL)
0.001 783: 50 mmol/Lチオ硫酸ナトリウム溶液1 mLのよう素酸カ
リウム相当量(g)
k) 10 mmol/Lチオ硫酸ナトリウム溶液 50 mmol/Lチオ硫酸ナトリウム溶液50 mLを全量フラスコ250
mLにとり,4.7 b) の溶存酸素を含まない水を標線まで加える。使用時に調製する。この溶液のファ
クタは,50 mmol/Lチオ硫酸ナトリウム溶液のものを用いる。
16.2.2
器具及び装置
器具及び装置は,次による。
a) 試料採取器 図15に示すような容量100±1 mLのもの2個。
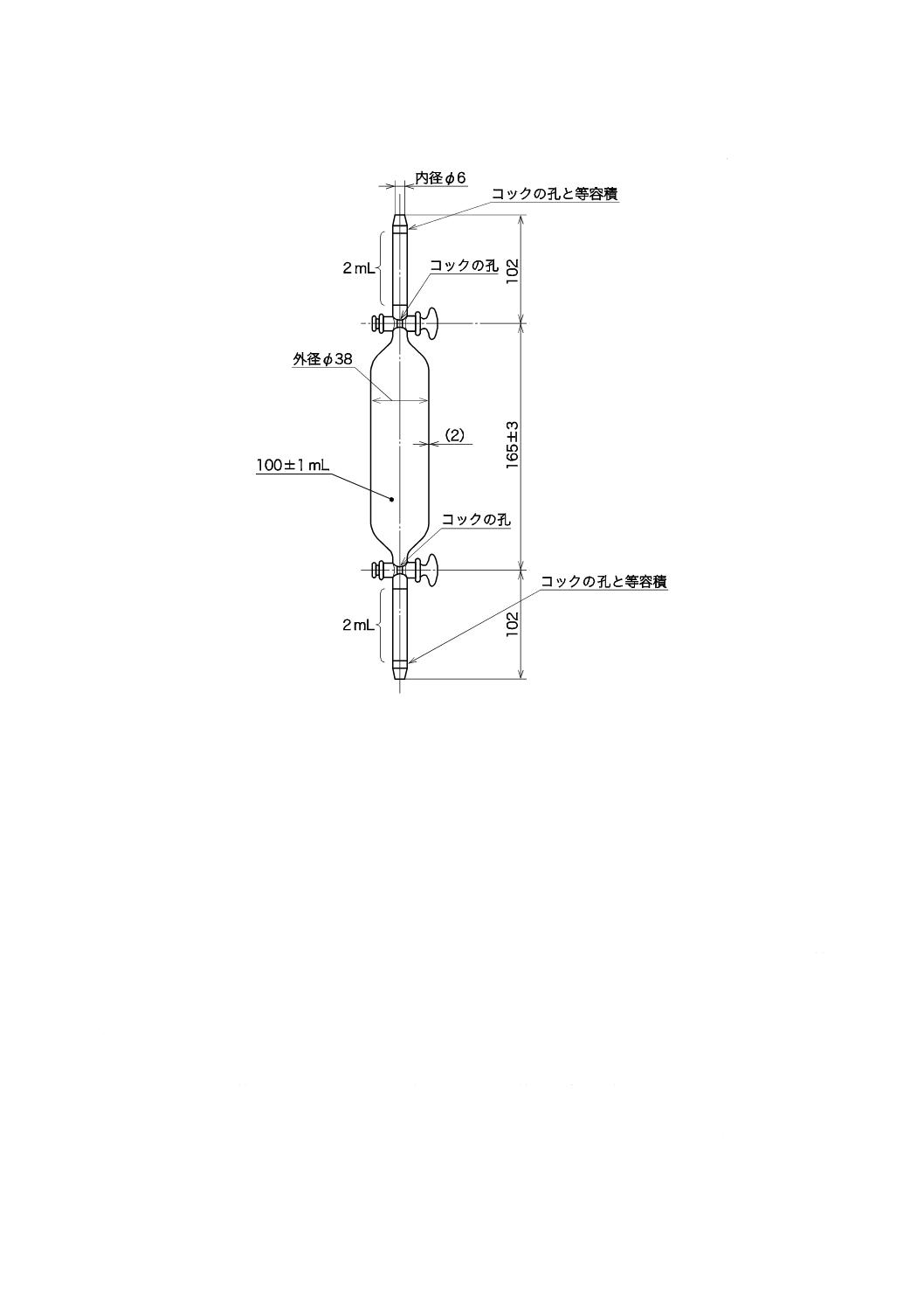
61
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
単位 mm
図15−試料採取器の一例
16.2.3
試料採取
試料採取は,次による。
a) 2個の試料採取器の出口を上方に向け,試料採取器の入口を配管の試料採取口よりも高い位置に支持
できるように組み立て,試料採取器の下端を空気が浸透しにくい導管とY字管とで試料採取口に接続
する(試料採取器の出口には何も接続しない。)。
注記 通常,使用する各種の導管のうち空気の浸透の少ないものは,軟質塩化ビニル,硬質ポリエ
チレン管,肉厚ゴム管などがある。
b) 試料の温度が室温よりも高い場合には,試料の温度が室温より1〜2 ℃低くなるように試料採取配管
中に適切な冷却蛇管を設ける。この場合には,冷却水調節用弁を冷却蛇管の入口に設けて冷却水を流
してあふれさせ,試料の流量調節弁は冷却蛇管の出口に設ける。
c) 2個の試料採取器とも同時に8〜12秒間で満たされるように試料の流量を調節し,試料配管中の滞留
水(元の試料)が完全に入れ代わるように,連続して試料を十分に流す。
なお,試料配管を断続的に使用する場合にも,試料配管及び冷却蛇管中の滞留水(元の試料)を完
全に置換するのに必要な時間だけ試料を流す。
d) 2個の試料採取器の上方のコックを閉じ,直ちに2個とも下方のコックを閉じ,接続管を外し,試料
採取器を逆にして気泡が全くないことを確かめる。ただし,少しでも気泡があれば,2個とも試料を
62
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
とり直す。
e) 試料採取器の1個を試験用,他を空試験用にする。
16.2.4
操作
操作は,次による。
a) 磁器蒸発皿によう素溶液(5 mmol/L)20 mLをとり,これに酢酸-酢酸ナトリウム緩衝液(pH 3.9)10 mL
を加える。
b) 試験用の試料採取器の両端にある足の部分の試料を捨てて水で洗浄し,両方の足にエタノール(95)2
mLずつを満たす。
c) 試料採取器の下方のエタノールをできるだけ捨てないようにして,磁器蒸発皿の溶液中に試料採取器
の先端を入れて上方のコックを開き,静かに下方のコックを開いて試料を注入する。
d) 10 mmol/Lチオ硫酸ナトリウム溶液で滴定し,よう素の黄色が薄くなったら,指示薬としてでんぷん
溶液(10 g/L)約1 mLを加え,生じたよう素でんぷんの青い色が消えるまで滴定する。
e) 空試験として,空試験用試料採取器から試料を三角フラスコ200 mLに移し入れ,硫酸(1+35)6〜7
mLを加え,窒素を液面に通しながら数分間静かに煮沸して二酸化硫黄を追い出す。窒素を通したま
ま冷却する。
f)
冷却後,指示薬としてフェノールフタレイン溶液(5 g/L)3〜5滴を加え,水酸化ナトリウム溶液(40
g/L)で中和する。これに,酢酸-酢酸ナトリウム緩衝液(pH 3.9)10 mLを加え,静かに振り混ぜた後,
よう素溶液(5 mmol/L)20 mLを加えd) の操作に従って,10 mmol/Lチオ硫酸ナトリウム溶液で滴定
する。
g) 次の式によって試料中の亜硫酸イオンの濃度(SO32−:mg/L)を算出する。
(
)
3
400
.0
000
1
×
×
×
−
=
V
f
b
a
S
ここに,
S: 亜硫酸イオン(SO32−:mg/L)
a: 滴定に要した10 mmol/Lチオ硫酸ナトリウム溶液(mL)
b: 空試験に要した10 mmol/Lチオ硫酸ナトリウム溶液
(mL)
f: 10 mmol/Lチオ硫酸ナトリウム溶液のファクタ
V: 試料(mL)
0.400 3: 10 mmol/Lチオ硫酸ナトリウム溶液1 mLの亜硫酸イオ
ン相当量(mg)
16.2.5
留意事項
a) 硫化物イオンは,よう素を消費して妨害し,16.2.4 e) の空試験を行っても補正されない。硫化物イオ
ンの妨害を除くには,次による。
1) JIS K 8953で規定する硫酸亜鉛七水和物20 gを水100 mLに溶かした溶液と炭酸ナトリウム溶液
(100 g/L)とを用意し,使用時にその等体積ずつを混合して塩基性炭酸亜鉛の懸濁液を調製する。
塩基性炭酸亜鉛の懸濁液10 mLは,硫化物イオン約50 mgを固定できる。
2) 図16の培養瓶に気泡が残らないように試料を採取する。
3) 塩基性炭酸亜鉛の懸濁液を試料100 mLにつき約2 mLの割合で試料の液面下に加える。
4) 気泡が残らないように注意して密栓し,転倒して混ぜ合わせて硫化物イオンを硫化亜鉛として沈殿
固定させる。
5) ろ紙5種Cでろ過(又は遠心分離)し,そのろ液を用いて16.2.4の操作を行って亜硫酸イオンを定

63
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
量する。
図16−培養瓶の例
b) 鉄(III)イオン及び銅(II)イオンはよう化物イオンを酸化して妨害する。
17
硫酸イオン(SO42−)
17.1
一般事項
硫酸イオンの定量には,硫酸バリウム比濁法,重量法,イオンクロマトグラフ法又は流れ分析法を適用
する。
17.2
硫酸バリウム比濁法
試料に塩化バリウムとコロイド安定剤とを加え,硫酸イオンとバリウムイオンとを反応させて硫酸バリ
ウムのコロイドとし,このコロイドの散乱による透過光の減少を見かけの吸光度として測定し,硫酸イオ
ンを定量する。
定量範囲:SO42− 20〜100 mg/L,繰返し精度:10 %
17.2.1
試薬
試薬は,次による。
a) 水 JIS K 0557で規定するA2又はA3の水。
b) 塩酸(1+50) JIS K 8180で規定する塩酸を用いて調製する。
c) 水酸化ナトリウム溶液(1 mol/L) JIS K 8576で規定する水酸化ナトリウム4 gを水に溶かして100 mL
とする。
d) グリセリン溶液(1+1) JIS K 8295で規定するグリセリンを用いて調製する。
e) 塩酸酸性塩化ナトリウム溶液 JIS K 8150で規定する塩化ナトリウム240 gをJIS K 8180で規定する
塩酸20 mLと水に溶かして1 Lとする。
f)
塩化バリウム JIS K 8155で規定する塩化バリウム二水和物をふるい分け,目開き710 μmのふるい
を通り,500 μmのふるいに止まるもの。
g) 硫酸イオン標準液(SO42−:1 000 mg/L) 国家計量標準にトレーサブルな硫酸イオン標準液(SO42−
1 000 mg/L)を使用するか,又は,次による。JIS K 8962で規定する硫酸カリウムを約700 ℃で約30
分間加熱し,デシケータ中で放冷する。その1.815 gをとり,少量の水に溶かして全量フラスコ1 000 mL
に移し入れ,水を標線まで加える。使用時に調製する。
64
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
17.2.2
器具及び装置
器具及び装置は,次による。
a) 光度計 分光光度計又は光電光度計。
b) マグネチックスターラ
17.2.3
操作
操作は,次による。
a) 5.3によってろ過した試料の50 mLを2個のコニカルビーカ100 mLにとる。硫酸イオン濃度が100 mg/L
以上の場合は,試料の適量をとり,水を加えて50 mLとする。
b) 試料が酸性又はアルカリ性の試料の場合は,塩酸(1+50)又は水酸化ナトリウム溶液(1 mol/L)を
用いてpHを約7に調節する。
c) それぞれにグリセリン溶液(1+1)10 mLと塩酸酸性塩化ナトリウム溶液5 mLとを加え,マグネチ
ックスターラを用いてかき混ぜる。このときのpHは1.4〜1.6になる。
d) 一方のコニカルビーカに塩化バリウム0.3 gを加え,それぞれ1分間引き続いてかき混ぜた後,4分間
放置し,再び15 秒間かき混ぜる。
e) 直ちにこの液を吸収セルに移し,塩化バリウムを加えない方を対照液とし,1分間以内に波長450 nm
付近の見かけの吸光度を測定する。
f)
空試験として水50 mLずつを2個のコニカルビーカにとり,c)〜e) の操作を行って試料について得た
吸光度を補正する。
g) あらかじめ,次によって作成した硫酸イオンの検量線から,試料中の硫酸イオンの濃度(SO42−:mg/L)
を算出する。a) で試料を適量とり,水で50 mLにした場合は,希釈率を考慮して濃度を算出する。
1) 硫酸イオン標準液(SO42−:1 000 mg/L)1〜5 mLを段階的にとり,水を加えて50 mLとし,c)〜f) の
操作を行って硫酸イオンの濃度(SO42−:mg/L)と見かけの吸光度との関係線を作成する。
17.3
重量法
重量法は,硫酸イオンを硫酸バリウムとして沈殿させ,その質量をはかって定量する。
定量範囲:SO42− 100 mg/L以上,繰返し精度:2 %
17.3.1
試薬
試薬は,次による。
a) 塩酸 JIS K 8180で規定するもの。
b) 塩酸(1+50) 17.2.1 b) による。
c) 塩化バリウム溶液(100 g/L) JIS K 8155で規定する塩化バリウム二水和物11.7 gを水に溶かして100
mLとする。
d) 硝酸銀溶液(10 g/L) JIS K 8550で規定する硝酸銀1 gを水に溶かして100 mLとする。
17.3.2
操作
操作は,次による。
a) 5.3によってろ過した試料の適量(SO42−として100 mg/L以上を含む。)を磁器蒸発皿にとり,塩酸3 mL
を加えた後,沸騰水浴上で蒸発乾固し,更に約20分間加熱する。
b) 放冷後,塩酸2 mLで湿し,次に,温水20〜30 mLを加え,数分間加熱した後,ろ紙5種Bでろ過し,
塩酸(1+50)で数回洗う。
c) ろ液に水を加えて100 mLとし,水浴上で加熱し,絶えずかき混ぜながら,これに温塩化バリウム溶
液(100 g/L)を滴加し,沈殿が生じなくなったら,更にその添加量の20〜50 %を過剰に加える。
65
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
d) 沸騰水浴上で20〜30分間加熱した後,3〜4時間放置する。
e) ろ紙6種又は5種Cを用いてろ過し,ろ液に塩化物イオンの反応を認めなくなるまで水で洗う[硝酸
銀溶液(10 g/L)で確かめる。]。
f)
沈殿はろ紙とともに,あらかじめ800 ℃で恒量とした磁器るつぼに入れ,乾燥後,徐々に加熱してろ
紙を,一旦,炭化した後,灰化する。
g) 引き続き,800 ℃で約30分間強熱し,デシケータ中で放冷した後,その質量をはかる。
h) g) の操作を繰り返して恒量とする。
i)
次の式によって試料中の硫酸イオンの濃度(SO42−:mg/L)を算出する。
(
)
6
411
.0
000
1
×
×
−
=
V
b
a
S
ここに,
S: 硫酸イオン(SO42−:mg/L)
a: 試験における磁器るつぼの質量(mg)
b: 空試験における磁器るつぼの質量(mg)
V: 試料(mL)
0.411 6: 硫酸バリウム1 mgの硫酸イオン相当量(mg)
17.4
イオンクロマトグラフ法
イオンクロマトグラフ法は,15.6による。
17.5
流れ分析法
陽イオン交換樹脂カラムを通して陽イオンを除去した試料の流れに,塩酸酸性バリウム・メチルチモー
ルブルーエタノール混合溶液の流れを混合すると,硫酸イオンがバリウムと反応して硫酸バリウムを生成
する。この流れに水酸化ナトリウム溶液の流れを混合すると,未反応のバリウムイオンとメチルチモール
ブルーとが錯体を生成し,溶液はその錯体による青い色と,未反応のメチルチモールブルーによる灰色を
呈する。未反応のメチルチモールブルーの灰色を460 nmで測定することによって硫酸イオンを定量する。
定量範囲:SO42− 2〜100 mg/L,繰返し精度:10 %以下(装置,測定条件によって異なる。)
17.5.1
メチルチモールブルー発色FIA法
17.5.1.1 試薬
試薬は,次による。調製した溶液は,必要に応じて脱気を行う。
a) 水 JIS K 0557で規定するA3又はA4の水。
b) キャリヤ液 硫酸イオン標準液(SO42−:1 000 mg/L)0.3 mLとり,水で1 Lとする。
c) 塩化バリウム溶液 JIS K 8155で規定する塩化バリウム二水和物1.526 gを約500 mLの水に溶かした
後,水で1 000 mLとし,ポリエチレン瓶で保存する。
d) 1.0 mol/L塩酸 JIS K 8180で規定する塩酸10 mLとり,水で120 mLとする。
e) 塩酸酸性バリウム・メチルチモールブルーエタノール混合溶液 メチルチモールブルー四ナトリウム
塩0.236 gに塩化バリウム溶液50 mLを加えてかき混ぜ溶解する。これに塩酸(1 mol/L)4 mLを加え
てかき混ぜ,70 mLの水と320 mLのエタノールを加えてかき混ぜる。褐色のポリ瓶に入れ冷蔵保存
する。
f)
水酸化ナトリウム原液 JIS K 8576で規定する水酸化ナトリウム25 gに水約40 mLを加えて溶解し,
水で全量を50 mLとする。
g) 水酸化ナトリウム溶液(0.18 mol/L) 水酸化ナトリウム原液9.9 gに水500 mLを加えて調製する。
h) 陽イオン交換カラム 50〜100メッシュの陽イオン交換樹脂0.5 gを,十分な水で混合することによっ
てスラリを作製し,気泡が入らないようにカラム充塡する。
66
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
i)
硫酸イオン標準液(SO42−:1 000 mg/L) 17.2.1 g) による。
17.5.1.2 器具及び装置
装置の基本構成は,次による(図17参照)。
a) 送液部 15.7.1.2 a) による。
b) 試料導入部 15.7.1.2 b) による。
c) 反応部 15.7.1.2 c) による。
d) 検出部 波長460 nm付近での測定が可能な吸光光度検出器を用いる。
e) 記録部 15.7.1.2 e) による。
C: キャリヤ液
R1: 塩酸酸性バリウム・メチルチモールブルーエタノール混合溶液
R2: 水酸化ナトリウム溶液(0.18 mol/L)
S: 試料
1: ポンプ(mL/min)
2: 試料導入部(試料200 μL)
3: 陽イオン交換カラム
4: 反応コイル(内径0.5 mm,長さ1 m)
5: 反応コイル(内径0.5 mm,長さ7 m)
6: 検出器(波長460 nm)
7: 廃液
図17−メチルチモールブルー発色FIA法のシステム例
17.5.1.3 準備操作
準備操作は,15.7.1.3による。ただし,感度の調節は硫酸イオンによる。
17.5.1.4 操作
操作は,15.7.1.4による。ただし,検量線は,17.5.1.1 i) を水で希釈して調製した検量線用硫酸イオン標
準液を用いて作成し,試料中の硫酸イオンの濃度の算出には,これを用いる。
17.5.1.5 留意事項
a) 多くの陽イオンが干渉するので,干渉を除去する陽イオン交換カラムを利用する。
b) モリブデンは僅か1 mg/Lで正の誤差を与える。
c) カラムが消耗すると試料中の陽イオンが未反応のメチルチモールブルーを消費するため,硫酸イオン
濃度は低い値を示す。この場合は新しい樹脂を再充塡する。
67
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
d) a)〜c) のほかは,15.7.1.5による。
17.5.2
メチルチモールブルー発色CFA法
17.5.2.1 試薬
試薬は,次による。調製した溶液は,必要に応じて脱気を行う。
a) 水 17.5.1.1 a) による。
b) 塩化バリウム溶液 17.5.1.1 c) による。
c) 塩酸酸性バリウム・メチルチモールブルーエタノール混合溶液 塩化バリウム溶液25 mLにメチルチ
モールブルー四ナトリウム塩を118.2 mg溶かした後,塩酸(1 mol/L)4 mLと70 mLの水とを加え,
JIS K 8102で規定するエタノール(95)で500 mLにする。使用時に調製する。
d) 水酸化ナトリウム溶液 JIS K 8576で規定する水酸化ナトリウム7.2 gを水に溶かして500 mLとする。
e) 硫酸イオン標準液(SO42−:1 000 mg/L) 17.2.1 g) による。
f)
ポリオキシエチレンドデシルエーテル溶液(300 g/L) ポリオキシエチレンドデシルエーテル30 g
をJIS K 8102に規定するエタノール(95)に溶かして100 mLとする。
g) 試料希釈水 硫酸イオン標準液(SO42−:1 000 mg/L)1 mLとポリオキシエチレンドデシルエーテル
溶液(300 g/L)1 mLとをとり,水で1 Lとする。
h) 陽イオン交換カラム 17.5.1.1 h) による。
i)
硫酸イオン標準液(SO42−:1 000 mg/L) 17.2.1 g) による。
17.5.2.2 器具及び装置
装置の基本構成は,次による(図18参照)。
a) 送液部 15.7.2.2 a) による。
b) 試料導入部 15.7.2.2 b) による。
c) 反応部 15.7.2.2 c) による。
d) 検出部 波長460 nm付近での測定が可能な吸光光度検出器を用いる。
e) 記録部 15.7.2.2 e) による。
68
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
S: 試料又は水
R1: 試料希釈水
R2: 塩酸酸性バリウム・メチルチモールブルーエタノール混合溶液
R3: 水酸化ナトリウム溶液
1: ポンプ(mL/min)
2: セグメントガス(空気)
3: 陽イオン交換カラム
4: 反応コイル(内径2 mm,長さ30 cm)
5: 反応コイル(内径2 mm,長さ120 cm)
6: 検出器(波長460 nm)
7: 廃液
図18−メチルチーモールブルー発色CFA法のシステム例
17.5.2.3 準備操作
準備操作は,15.7.2.3による。ただし,感度の調節は硫酸イオンによる。
17.5.2.4 操作
操作は,15.7.2.4による。ただし,検量線は,17.5.2.1 i) を水で希釈して調製した検量線用硫酸イオン標
準液を用いて作成し,試料中の硫酸イオンの濃度の算出には,これを用いる。
17.5.2.5 留意事項
留意事項は,17.5.1.5のa)〜c) 及び15.7.2.5による。
18
りん酸イオン(PO43−)及び加水分解性りん酸イオン
18.1
一般事項
ボイラ水及び給水中の溶存状態のオルトりんイオン及びメタりん酸イオン,トリポリりん酸イオンなど
の加水分解性のりん酸イオンだけを定量する。懸濁物を含む試料の場合には,懸濁物を除去する前処理を
69
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
行った後,定量する。
18.2
りん酸イオン
りん酸イオンの定量には,モリブデン青[アスコルビン酸還元]吸光光度法を用いる。
18.2.1
モリブデン青[アスコルビン酸還元]吸光光度法
りん酸イオンが七モリブデン酸六アンモニウム及びタルトラトアンチモン(III)酸カリウムと反応して
生成するヘテロポリ化合物をL(+)-アスコルビン酸で還元し,生成したモリブデン青の吸光度を測定して
りん酸イオンを定量する。
定量範囲:PO43− 0.1〜3 mg/L,繰返し精度:2〜10 %
18.2.1.1 試薬
試薬は,次による。
a) 水 JIS K 0557で規定するA3の水。
b) アスコルビン酸溶液(72 g/L) JIS K 9502で規定するL(+)-アスコルビン酸7.2 gを水に溶かして100
mLとする。0〜10 ℃の暗所に保存し,着色した溶液は使用しない。
c) モリブデン酸アンモニウム溶液 JIS K 8905で規定する七モリブデン酸六アンモニウム四水和物6 g
及びJIS K 8533で規定するビス[(+)-タルトラト]二アンチモン(III)酸二カリウム三水和物0.24 g
を水約300 mLに溶かし,これに硫酸(2+1)(JIS K 8951で規定する硫酸を用いて調製する。)120 mL
を加え,次に,JIS K 8588で規定するアミド硫酸アンモニウム5 gを加えて溶かした後,水を加えて
500 mLとする。
d) モリブデン酸アンモニウム-アスコルビン酸混合溶液 モリブデン酸アンモニウム溶液とアスコルビ
ン酸溶液(72 g/L)とを体積比で5:1の割合になるように混合する。使用時に調製する。
e) p-ニトロフェノール溶液(1 g/L) JIS K 8721で規定するp-ニトロフェノール0.1 gを水に溶かして
100 mLとする。
f)
りん酸イオン標準液(PO43−:1 000 mg/L) 国家計量標準にトレーサブルなりん酸イオン標準液
(PO43 −:1 000 mg/L)を使用するか,又は,次による。JIS K 9007で規定するりん酸二水素カリウム
(pH標準液用)を105±2 ℃で約2時間加熱し,デシケータ中で放冷する。その1.433 gをとり,少量
の水に溶かして全量フラスコ1 000 mLに移し入れ,水を標線まで加える。0〜10 ℃の暗所に保存する。
g) りん酸イオン標準液(PO43−:100 mg/L) りん酸イオン標準液(PO43−:1 000 mg/L)10 mLを全量
フラスコ100 mLにとり,水を標線まで加える。0〜10 ℃の暗所に保存する。
h) りん酸イオン標準液(PO43−:5 mg/L) りん酸イオン標準液(PO43−:100 mg/L)10 mLを全量フラ
スコ200 mLにとり,水を標線まで加える。
i)
硫酸(1+50) JIS K 8951で規定する硫酸を用いて調製する。
j)
水酸化ナトリウム溶液(1 mol/L) JIS K 8576で規定する水酸化ナトリウム4 gを水に溶かして100 mL
とする。
18.2.1.2 器具及び装置
器具及び装置は,次による。
a) 光度計 分光光度計又は光電光度計。
b) メスシリンダ(有栓形) 15.2.2 b) による。
18.2.1.3 操作
操作は,次による。
a) 5.3によってろ過した試料25 mLをメスシリンダ(有栓形)25 mLにとる。りん酸イオン濃度が3 mg/L
70
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
以上の場合は,試料の適量をとり,水で25 mLとする。
b) 試料が酸性又はアルカリ性の場合は,指示薬としてp-ニトロフェノール溶液(1 g/L)1,2滴を加え,
硫酸(1+50)又は水酸化ナトリウム溶液(1 mol/L)を用いて僅かに黄に呈色するまで中和して一定
量とした試料を用いる。
c) モリブデン酸アンモニウム-アスコルビン酸混合溶液2 mLを加えて振り混ぜた後,20〜40 ℃で約15
分間放置する。
d) 溶液の一部を吸収セルに移し,波長880 nm又は710 nm付近の吸光度を測定する。
e) 空試験として水25 mLをとり,c) 及びd) の操作を行って吸光度を測定し,試料について得た吸光度
を補正する。
f)
あらかじめ,次によって作成したりん酸イオンの検量線から,試料中のりん酸イオンの濃度(PO43−:
mg/L)を算出する。a) で試料の適量をとり,水で希釈した場合は,希釈率を考慮して濃度を算出する。
1) りん酸イオン標準液(PO43−:5 mg/L)0.5〜15 mLをメスシリンダ(有栓形)25 mLに段階的にとり
水で25 mLとする。c) 及びd) の操作を行って吸光度を測定し,りん酸イオン(PO43−)の濃度と
吸光度との関係線を作成する。発色操作は試料と同じ温度で行う。
18.2.1.4 留意事項
a) ろ過しても濁りや色がある場合は,モリブデン酸アンモニウム-アスコルビン酸混合溶液2 mLの代わ
りに,モリブデン酸アンモニウム溶液2 mLを添加し,これを対照液とする。
b) 硝酸イオンや亜硝酸イオンが試料に存在すると,生成したモリブデン青が退色するなどの妨害がある
が,アミド硫酸アンモニウムを添加すれば,この妨害を防ぐことができる。
c) 鉄(III)30 mg以上の共存もモリブデン青を退色させるが,アスコルビン酸溶液(72 g/L)の添加量を
増せば,妨害を抑制できる。
d) 試料中にひ素(V)が含まれるときは,次の操作を行って定量値に正の誤差を与える妨害を除去する。
1) ろ過した試料20 mLに硫酸(1 mol/L)(JIS K 8951で規定する硫酸を用いて調製する。)1 mLとチ
オ硫酸ナトリウム溶液(JIS K 8637で規定するチオ硫酸ナトリウム五水和物1.2 gとを水に溶かして
100 mLとし,JIS K 8625で規定する炭酸ナトリウム50 mgを加えて調製する。)0.5 mLを加え,5
〜10分間放置する。18.2.1.3 b) によって中和し,水で25 mLにする。
e) 次のような操作で発色させ,定量することもできる。
1) 5.3によってろ過した試料40 mLを全量フラスコ50 mLにとる。りん酸イオン濃度が高い場合は,
試料の適量をとり,水を加えて約40 mLとする。
2) 試料が酸性又はアルカリ性の試料の場合は,指示薬としてp-ニトロフェノール溶液(1 g/L)1,2
滴を加え,硫酸(1+50)又は水酸化ナトリウム溶液(1 mol/L)を用いて僅かに黄色を呈色するま
で中和して一定量とした試料を用いる。
3) モリブデン酸アンモニウム-アスコルビン酸混合溶液3.5 mLを加え,水を標線まで加えて振り混ぜ
た後,20〜40 ℃で約15分間放置する。
4) 溶液の一部を吸収セルに移し,波長880 nm又は710 nm付近の吸光度を測定する。
5) 空試験として水約40 mLを全量フラスコ50 mLにとり,3) 及び4) の操作を行って吸光度を測定し,
試料について得た吸光度を補正する。
6) 検量線からりん酸イオンの濃度を求め,試料中のりん酸イオンの濃度(PO43−:mg/L)を算出する。
18.2.2
流れ分析法
試料中のりん酸イオンを,18.2.1と同様な原理で発色させる流れ分析法によって定量する。
71
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
定量範囲:PO43− 0.03〜3 mg/L,繰返し精度:10 %以下(装置,測定条件によって異なる。)
18.2.2.1 モリブデン青発色3流路FIA法
18.2.2.1.1 試薬
試薬は,次による。調製した溶液は,必要に応じて脱気を行う。
a) 水 18.2.1.1 a) による。
b) モリブデン酸アンモニウム溶液 JIS K 8905で規定する七モリブデン酸六アンモニウム四水和物40 g
を水約800 mLに加えて溶かした後,水で1 000 mLとする。
c) タルトラトアンチモン(III)酸カリウム溶液 JIS K 8533で規定するビス[(+)-タルトラト]二アン
チモン(III)酸二カリウム三水和物3.0 gを水約800 mLに加えて溶かした後,水で1 000 mLとする。
d) 硫酸酸性(0.63 mol/L)-モリブデン酸アンモニウム溶液 水約500 mLをビーカにとり,これをかき
混ぜながら,JIS K 8951で規定する硫酸35 mLを徐々に加える。冷却後,モリブデン酸アンモニウム
溶液213 mLとタルトラトアンチモン(III)酸カリウム溶液72 mLとを加え,全量を水で1 000 mLと
する。
e) アスコルビン酸溶液(60 g/L) JIS K 9502で規定するL(+)-アスコルビン酸6.0 gを水約80 mLに加
えて溶かした後,ドデシル硫酸ナトリウム0.1 gを加えて,全量を水で100 mLとする。この溶液は,
使用前に調製する。
f)
キャリヤ液 水を用いる。
g) りん酸イオン標準液(PO43−:100 mg/L) 18.2.1.1 g) による。
h) りん酸イオン標準液(PO43−:30 mg/L) 全量フラスコ100 mLにりん酸イオン標準液(PO43−:100 mg/L)
30 mLをとり,水を標線まで加える。使用時に調製する。
i)
りん酸イオン標準液(PO43−:10 mg/L) 全量フラスコ100 mLにりん酸イオン標準液(PO43−:100 mg/L)
10 mLをとり,水を標線まで加える。使用時に調製する。
18.2.2.1.2 器具及び装置
装置の基本構成は,次による(図19参照)。
a) 送液部 15.7.1.2 a) による。
b) 試料導入部 15.7.1.2 b) による。
c) 反応部 15.7.1.2 c) 及び一定の温度に加熱保持できる恒温槽から構成する。
d) 検出部 波長880 nm付近での測定が可能な吸光光度検出器を用いる。
e) 記録部 15.7.1.2 e) による。
f)
恒温槽 60 ℃に加熱できるもの。
72
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
C: キャリヤ液
R1: 硫酸酸性(0.63 mol/L)-モリブデン酸アンモニウム溶液
R2: アスコルビン酸溶液(60 g/L)
S: 試料
1: ポンプ(mL/min)
2: 試料導入器[注入量640 μL(りん酸イオン濃度0.01〜0.10 mg/L),注入量100 μL(りん酸イオン濃度0.1〜
1.0 mg/L)]
3: 反応コイル(内径0.7 mm,長さ120 cm)
4: 恒温槽(60 ℃)
5: 反応コイル(内径0.7 mm,長さ150 cm)
6: 検出器(波長880 nm)
7: 廃液
図19−モリブデン青発色3流路FIA法のシステム例
18.2.2.1.3 準備操作
準備操作は,15.7.1.3による。ただし,感度の調節はりん酸イオンによる。
18.2.2.1.4 操作
操作は,15.7.1.4による。ただし,検量線は,18.2.2.1.1 h) 又はi) を水で希釈して調製した検量線用りん
酸イオン標準液を用いて作成し,試料中のりん酸イオンの濃度の算出には,これを用いる。
18.2.2.1.5 留意事項
a) ひ素イオン,亜硝酸イオン及び酸化性物質は定量を妨害する。妨害物質の詳細については,18.2.1.4
による。
b) a) のほかは,15.7.1.5による。
18.2.2.2 モリブデン青発色2流路FIA法
18.2.2.2.1 試薬
試薬は,次による。調製した溶液は,必要に応じて脱気を行う。
a) 水 18.2.1.1 a) による。
b) 硫酸酸性(1.21 mol/L)-モリブデン酸アンモニウム溶液 水約800 mLをビーカにとり,これをかき
混ぜながら,JIS K 8951で規定する硫酸67 mLを徐々に加える。これにJIS K 8905で規定する七モリ
ブデン酸六アンモニウム四水和物5.48 gとJIS K 8533で規定するビス[(+)-タルトラト]二アンチモ
ン(III)酸二カリウム三水和物0.25 gとを加えて溶かした後,水で1 000 mLとする。
c) アスコルビン酸溶液(3 g/L) JIS K 9502で規定するL(+)-アスコルビン酸3.0 gとドデシル硫酸ナ
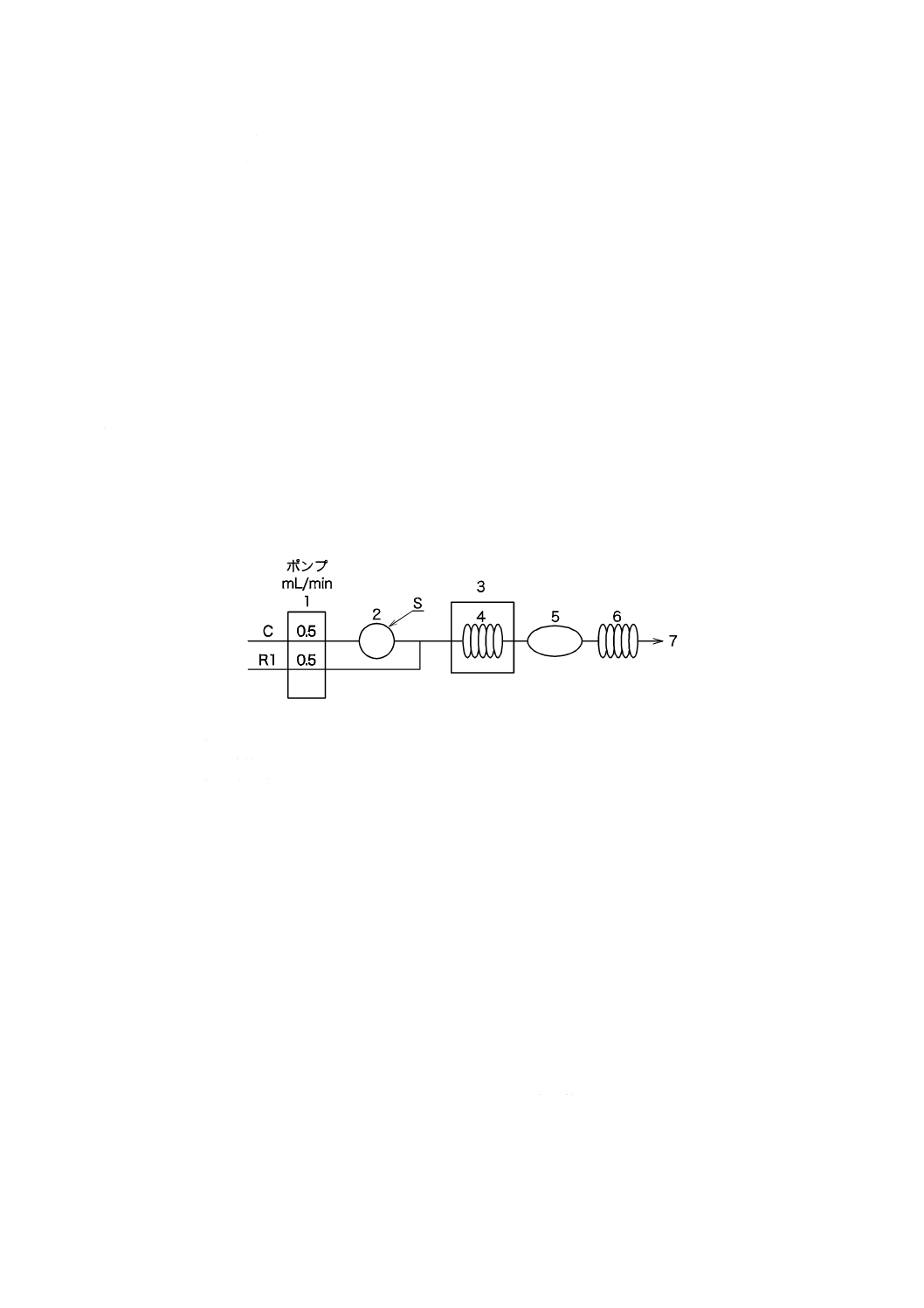
73
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
トリウム1 gとを水1 000 mLに溶かす。この溶液は,使用前に調製する。
d) 発色試薬溶液 硫酸酸性(1.21 moL/L)-モリブデン酸アンモニウム溶液とアスコルビン酸溶液(3 g/L)
とを等量混合する。この溶液は,使用前に調製する。
e) キャリヤ液 水を用いる。
f)
りん酸イオン標準液(PO43−:100 mg/L) 18.2.1.1 g) による。
g) りん酸イオン標準液(PO43−:30 mg/L) 18.2.2.1.1 h) による。
h) りん酸イオン標準液(PO43−:10 mg/L) 18.2.2.1.1 i) による。
18.2.2.2.2 器具及び装置
装置の基本構成は,次による(図20参照)。
a) 送液部 15.7.1.2 a) による。
b) 試料導入部 15.7.1.2 b) による。
c) 反応部 15.7.1.2 c) 及び一定の温度に加熱保持できる恒温槽から構成する。
d) 検出部 18.2.2.1.2 d) による。
e) 記録部 15.7.1.2 e) による。
f)
恒温槽 70 ℃に加熱できるもの。
C: キャリヤ液
R1: 発色試薬溶液
S: 試料
1: ポンプ(mL/min)
2: 試料導入部(試料300 μL)
3: 恒温槽(70 ℃)
4: 反応コイル(内径0.5 mm,長さ7 m)
5: 検出器(波長880 nm)
6: 背圧コイル(内径2 mm,長さ0.5 m)
7: 廃液
図20−モリブデン青発色2流路FIA法のシステム例
18.2.2.2.3 準備操作
準備操作は,15.7.1.3による。ただし,感度の調節はりん酸イオンによる。
18.2.2.2.4 操作
操作は,15.7.1.4による。ただし,検量線は,18.2.2.2.1 g) 又はh) を水で希釈して調製した検量線用り
ん酸イオン標準液を用いて作成し,試料中のりん酸イオンの濃度の算出には,これを用いる。
18.2.2.2.5 留意事項
留意事項は,18.2.2.1.5による。
74
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
18.2.2.3 モリブデン青発色CFA法
18.2.2.3.1 試薬
試薬は,次による。調製した溶液は,必要に応じて脱気を行う。
a) 水 18.2.2.1.1 a) による。
b) モリブデン酸アンモニウム溶液 18.2.2.1.1 b) による。
c) タルトラトアンチモン(III)酸カリウム溶液 JIS K 8533で規定するビス[(+)-タルトラト]二アン
チモン(III)酸二カリウム三水和物2.5 gを水約800 mLに加えて溶かした後,水で1 000 mLとする。
d) 硫酸(2.45 mol/L) 水約800 mLをビーカにとり,これをかき混ぜながら,JIS K 8951で規定する硫
酸135 mLを徐々に加える。冷却後,水を加えて全量を1 000 mLとする。
e) 硫酸酸性(1.75 mol/L)-モリブデン酸アンモニウム溶液 硫酸(2.45 mol/L)500 mLとモリブデン酸
アンモニウム溶液150 mLとタルトラトアンチモン(III)酸カリウム溶液50 mLとを混合する。
f)
アスコルビン酸溶液(10 g/L) JIS K 9502で規定するL(+)-アスコルビン酸1 gを水約80 mLに加え
て溶かした後,水で100 mLとする。使用時に調製する。
g) 洗剤溶液I ドデシル硫酸ナトリウム1 gを水約800 mLに溶かし,全量を水で1 000 mLとする。
h) 洗剤溶液II ドデシル硫酸ナトリウム10 gを水約800 mLに溶かし,全量を水で1 000 mLとする。
i)
りん酸イオン標準液(PO43−:100 mg/L) 18.2.1.1 g) による。
j)
りん酸イオン標準液(PO43−:30 mg/L) 18.2.2.1.1 h) による。
k) りん酸イオン標準液(PO43−:10 mg/L) 18.2.2.1.1 i) による。
18.2.2.3.2 器具及び装置
装置の基本構成は,次による(図21参照)。
a) 送液部 15.7.2.2 a) による。
b) 試料導入部 15.7.2.2 b) による。
c) 反応部 15.7.2.2 c) 及び一定の温度に加熱保持できる恒温槽から構成する。
d) 検出部 18.2.2.1.2 d) による。
e) 記録部 15.7.2.2 e) による。
f)
恒温槽 37〜40 ℃に加熱できるもの。
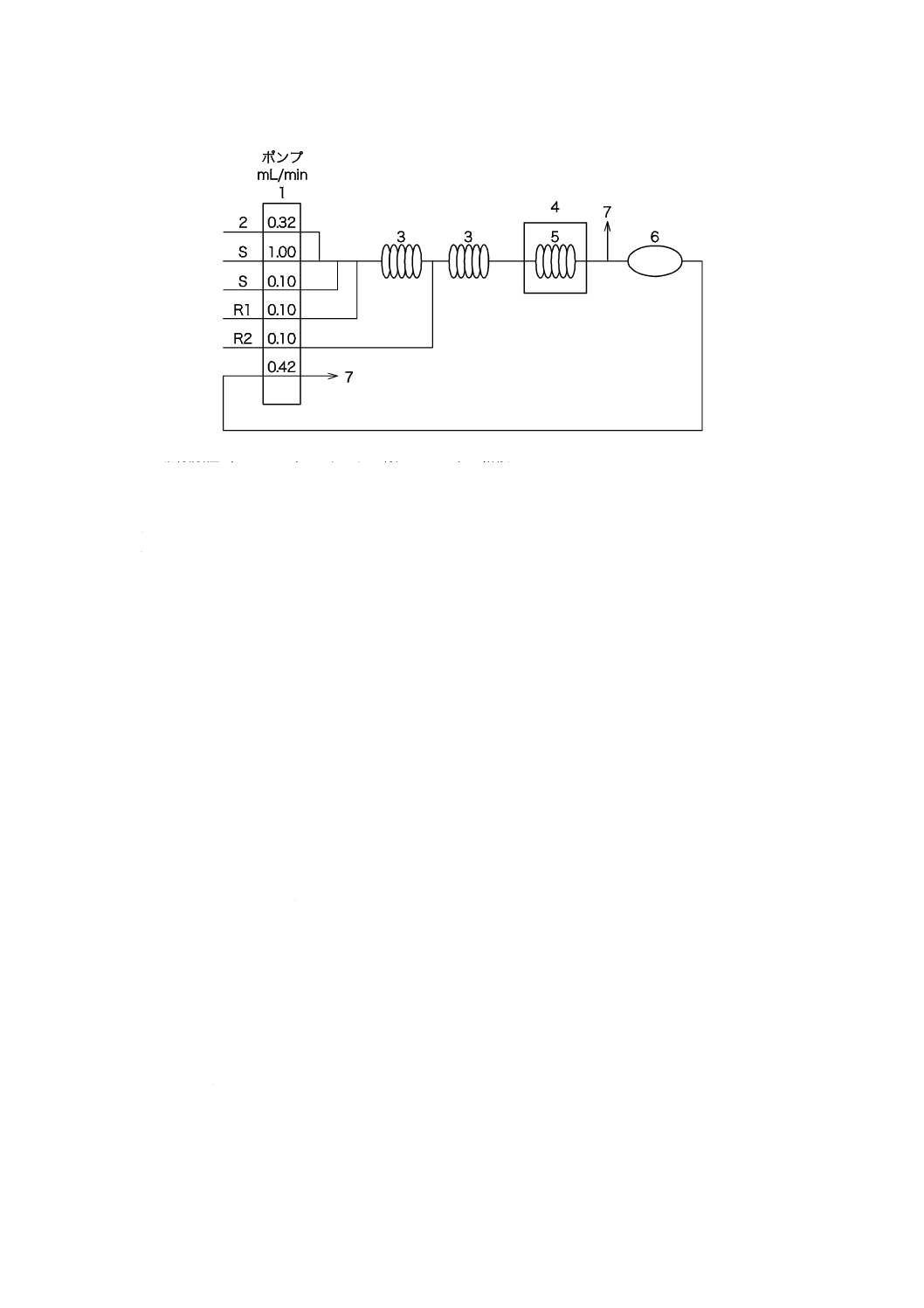
75
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
R1: 硫酸酸性(1.75 mol/L)-モリブデン酸アンモニウム溶液
R2: アスコルビン酸溶液(10 g/L)
S: 試料又は洗剤溶液[定量範囲0.01〜0.10 mg/Lの場合(試料流量1.00 mL/min,洗剤溶液II流量0.10 mL/min),
定量範囲0.1〜1.0 mg/Lの場合(試料流量0.1 mL/min,洗剤溶液I流量1.00 mL/min)]
1: ポンプ(mL/min)
2: セグメントガス(空気)
3: 反応コイル(内径2 mm,長さ30 cm)
4: 恒温槽(37〜40 ℃)
5: 加熱コイル(内径2 mm,長さ120 cm)
6: 検出器(波長880 nm)
7: 廃液
図21−モリブデン青発色CFA法のシステム例
18.2.2.3.3 準備操作
準備操作は,15.7.2.3による。ただし,感度の調節はりん酸イオンによる。
18.2.2.3.4 操作
操作は,15.7.2.4による。ただし,検量線は,18.2.2.3.1 j) 又はk) を水で希釈して調製した検量線用り
ん酸イオン標準液を用いて作成し,試料中のりん酸イオンの濃度の算出には,これを用いる。
18.2.2.3.5 留意事項
留意事項は,18.2.2.1.5 a) によるほかは,15.7.2.5による。
18.3
加水分解性りん酸イオン
試料を酸性として煮沸したとき,加水分解によってりん酸イオンとなるメタりん酸イオン,トリポリり
ん酸イオンなどをいう。
試料に硫酸-硝酸の混酸を加え,煮沸してりん酸イオンとした後,18.2.1,又は18.2.2によってりん酸イ
オン濃度を定量し,この値から加水分解前のりん酸イオン濃度を差し引き,加水分解性りん酸イオンをり
ん酸イオンに換算した濃度で表示する。
18.3.1
試料の前処理
試料を酸性として煮沸し,加水分解によってりん酸イオンにする。
18.3.1.1 試薬
試薬は,次による。
76
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) 水 JIS K 0557で規定するA3の水。
b) 硫酸-硝酸の混酸 JIS K 8951で規定する硫酸300 mLを水約600 mL中に注意してかき混ぜながら加
えた後,放冷する。これにJIS K 8541で規定する硝酸4 mLと水とを加えて全量を1 Lとする。
c) 水酸化ナトリウム溶液(40 g/L) JIS K 8576で規定する水酸化ナトリウム4 gを水に溶かして100 mL
とする。
d) p-ニトロフェノール溶液(1 g/L) 18.2.1.1 e) による。
18.3.1.2 操作
操作は,次による。
a) 5.3によってろ過した試料100 mLをビーカ200 mLにとり,硫酸-硝酸の混酸1 mLを加える。
b) 静かに煮沸する。液量が25 mL以下になったら水を加え,液量を25〜50 mLに保って,約90分間煮
沸する。二りん酸イオン,トリポリりん酸イオンなどは約1時間でりん酸イオンになる。
c) 放冷後,ろ紙5種Bを用いてろ過し,温水で3〜4回洗う。
d) ろ液及び洗液を合わせ,指示薬としてp-ニトロフェノール溶液(1 g/L)数滴を加え,溶液が僅かに黄
に呈色するまで水酸化ナトリウム溶液(40 g/L)を滴加した後,全量フラスコ100 mLに移し入れ,水
を標線まで加える。
18.3.2
モリブデン青[アスコルビン酸還元]吸光光度法
18.3.1で処理した試料のりん酸イオン濃度から,5.3によってろ過した試料のりん酸イオン濃度を差し引
いて,試料中の加水分解性りん酸イオン濃度(PO43−:mg/L)を求める。試薬,器具及び装置,操作は18.2.1
による。
18.3.3
流れ分析法
18.3.1で処理した試料のりん酸イオン濃度から,5.3によってろ過した試料のりん酸イオン濃度を差し引
いて,試料中の加水分解性りん酸イオン濃度(PO43−:mg/L)を求める。試薬,器具及び装置,操作は18.2.2
による。
19
シリカ(SiO2)
19.1
一般事項
水中のシリカはイオン状シリカ(イオン状けい酸),溶存及びコロイド状シリカ,及び全シリカに区分し,
いずれも酸化けい素(IV)(SiO2)として表示する。
19.2
イオン状シリカ
イオン状シリカは七モリブデン酸六アンモニウムと反応してヘテロポリ化合物の黄色を生成するシリカ
をいう。イオン状シリカの定量にはモリブデン黄吸光光度法,モリブデン青吸光光度法,モリブデン青抽
出吸光光度法,流れ分析法又はプロセス用分析装置による測定方法を適用する。
19.2.1
モリブデン黄吸光光度法
イオン状シリカが,七モリブデン酸六アンモニウムと反応して生成するヘテロポリ化合物の黄色の吸光
度を測定してシリカを定量する。しゅう酸溶液を加えて試料中の共存りん酸イオンによって生成したヘテ
ロポリ化合物を分解する。
定量範囲:SiO2:2〜20 mg/L,繰返し精度:2〜10 %
19.2.1.1 試薬
試薬は,次による。ポリエチレン瓶に保存する。
a) 水 JIS K 0557で規定するA3の水。
77
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) 塩酸(1+1) JIS K 8180で規定する塩酸を用いて調製する。
c) モリブデン酸アンモニウム溶液(100 g/L) JIS K 8905で規定する七モリブデン酸六アンモニウム四
水和物21.2 gを水に溶かして200 mLとする。
d) しゅう酸溶液 JIS K 8519で規定するしゅう酸二水和物20 gを水に溶かして200 mLとする。
e) シリカ標準液(SiO2:1 000 mg/L) 砂状の石英(99.9 %以上)を,めのう乳鉢ですり潰し,700〜800 ℃
で約1時間加熱した後,デシケータ中で放冷する。その0.500 gを白金るつぼにとり,JIS K 8005で規
定する容量分析用標準物質の炭酸ナトリウム4 gを加え,十分に混合した後,加熱し,約40分間融解
する。放冷後,融成物を水に溶かして全量フラスコ500 mLに移し入れ,水を標線まで加える。
f)
シリカ標準液(SiO2:100 mg/L) シリカ標準液(SiO2:1 000 mg/L)20 mLを全量フラスコ200 mL
にとり,水を標線まで加える。
19.2.1.2 器具及び装置
器具及び装置は,次による。
a) 光度計 分光光度計又は光電光度計。
b) メスシリンダ(有栓形) 15.2.2 b) による。
19.2.1.3 操作
操作は,次による。
a) 5.3によってろ過した試料50 mLをメスシリンダ(有栓形)50 mLにとる。イオン状シリカ濃度が10
mg/L以上の場合は,試料の適量をとり,水を加えて50 mLとする。
b) 液温を約20 ℃に調節する。
c) 塩酸(1+1)1 mLとモリブデン酸アンモニウム溶液(100 g/L)2 mLとを加えて振り混ぜ,約5分間
放置する。
d) しゅう酸溶液1.5 mLを加えて振り混ぜ,1分間放置する。りん酸イオンが共存しない場合には,しゅ
う酸は添加しない。ただし,検量線の作成時も同様にする。
e) 直ちにこの溶液を吸収セルに移し,波長410〜450 nmの吸光度を測定する。
f)
空試験として水約50 mLをとり,b)〜e) の操作を行って吸光度を求め,試料について得た吸光度を補
正する。
g) あらかじめ,次によって作成したシリカの検量線から,試料中のシリカの濃度(SiO2:mg/L)を算出
する。
1) シリカ標準液(SiO2:100 mg/L)1〜10 mLをメスシリンダ(有栓形)50 mLに段階的にとり,水を
50 mLの標線まで加えた後,b)〜e) の操作を行ってシリカの濃度(SiO2:mg/L)と吸光度との関係
線を作成する。
19.2.1.4 留意事項
a) 試料が強いアルカリ性の場合には,塩酸(1+1)を用いて中和しておく。また,酸性の場合はモリブ
デン酸アンモニウム溶液を添加し,pHが1.1〜1.6になるように塩酸(1+1)の添加量を調節する。酸
性が強い場合には,水酸化ナトリウム溶液で中和する。
b) しゅう酸溶液を加えた場合には,放置時間を正しく守る。放置時間が長くなると,シリカによるヘテ
ロポリ化合物の黄色が退色する。
19.2.2
モリブデン青吸光光度法
イオン状シリカが七モリブデン酸六アンモニウムと反応して生成するヘテロポリ化合物をL(+)-アスコ
ルビン酸で還元してモリブデン青に変え,その吸光度を測定してシリカを定量する。
78
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
定量範囲: 10 mmセル使用の場合SiO2:0.2〜2 mg/L
20 mmセル使用の場合SiO2:0.1〜1 mg/L
50 mmセル使用の場合SiO2:20〜300 µg/L
繰返し精度:2〜10 %
19.2.2.1 試薬
試薬は,次による。ポリエチレン瓶に保存する。
a) 水 19.2.1.1 a) による。
b) 塩酸(1+1) 19.2.1.1 b) による。
c) 硫酸(1+5) 水5容をビーカにとり,これを冷却し,かき混ぜながらJIS K 8951で規定する硫酸1
容を徐々に加える。
d) モリブデン酸アンモニウム溶液(100 g/L) 19.2.1.1 c) による。
e) しゅう酸溶液 19.2.1.1 d) による。
f)
アスコルビン酸溶液(100 g/L) JIS K 9502で規定するL(+)-アスコルビン酸10 gを水に溶かして
100 mLとする。10 ℃以下の暗所に保存し,色の着いた溶液は使用しない。
g) シリカ標準液(SiO2:10 mg/L) 19.2.1.1 f) のシリカ標準液(SiO2:100 mg/L)20 mLを全量フラス
コ200 mLにとり,水を標線まで加える。使用時に調製する。
19.2.2.2 器具及び装置
器具及び装置は,次による。
a) 光度計 分光光度計又は光電光度計。
b) メスシリンダ(有栓形) 15.2.2 b) による。
19.2.2.3 操作
操作は,次による。
a) 5.3によってろ過した試料の50 mL(試料濃度としてSiO2:2 mg/L以下)をメスシリンダ(有栓形)
にとる。濃度が高い場合は,試料の適量をとり,水を加えて50 mLとする。
b) 液温を約20 ℃に調節する。
c) 塩酸(1+1)1 mL[又は硫酸(1+5)1 mL]とモリブデン酸アンモニウム溶液(100 g/L)2 mLとを
加えて振り混ぜ,溶液のpHを1.1〜1.6にし約5分間放置する。
d) しゅう酸溶液1.5 mLを加えて振り混ぜ,1分間放置する。
e) アスコルビン酸溶液(100 g/L)1 mLを加えて振り混ぜ,約10分間放置する。
f)
溶液の一部を吸収セルに移し,波長815 nm付近の吸光度を測定する。
g) 空試験として水50 mLをとり,b)〜f) の操作を行って吸光度を求め,試料について得た吸光度を補正
する。
h) あらかじめ,次によって作成したシリカの検量線から,試料中のシリカの濃度(SiO2:mg/L)を算出
する。a) で試料を適量とり,水で50 mLにした場合は,希釈率を考慮して濃度を算出する。
1) セル長が10 mmの場合,シリカ標準液(SiO2:10 mg/L)1〜10 mL,セル長が20 mmの場合,シリ
カ標準液(SiO2:10 mg/L)0.5〜5 mLをメスシリンダ(有栓形)50 mLに段階的にとり,水を50 mL
の標線まで加える。セル長が50 mmの場合には,シリカ標準液(SiO2:10 mg/L)1〜15 mLを全量
500 mLフラスコに段階的にとり,標線まで水を加え,そこから50 mLをメスシリンダ(有栓形)
50 mLにとる。液温を約20 ℃に調節し,b)〜f) の操作を行って吸光度を測定し,シリカの濃度
(SiO2:mg/L)と吸光度との関係を作成する。
79
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
19.2.2.4 留意事項
a) りん酸イオンが共存しなくてもしゅう酸溶液を加える。しゅう酸を加えないと,過剰のモリブデン酸
もアスコルビン酸溶液で還元され青い色となり妨害する。
19.2.3
モリブデン青抽出吸光光度法
イオン状シリカが七モリブデン酸六アンモニウムと反応して生成するヘテロポリ化合物をL(+)-アスコ
ルビン酸で還元してモリブデン青に変え,これを1-ブタノールに抽出し,有機層の吸光度を測定してシリ
カを定量する。この方法は,シリカの濃度の低い試料に適用する。
定量範囲: 10 mmセル使用の場合SiO2:10〜100 µg/L
20 mmセル使用の場合SiO2:2.5〜50 µg/L
繰返し精度:5〜20 %
19.2.3.1 試薬
試薬は,次による。ポリエチレン瓶に保存する。
a) 水 JIS K 0557で規定するA4の水。使用前に空試験を行い,使用の可否を確認する。
b) 硫酸(2.5 mol/L)-モリブデン酸アンモニウム(188 g/L)混合溶液 JIS K 8951で規定する硫酸140 mL
を水約300 mLに冷却しながら混合する。これにJIS K 8905で規定する七モリブデン酸六アンモニウ
ム四水和物200 gを水約500 mLに溶かした溶液を加え,全量フラスコ1 000 mLに移し入れ,水を標
線まで加える。
c) 硫酸(2+1) 水1容をビーカにとり,これを冷却し,かき混ぜながらJIS K 8951で規定する硫酸2
容を徐々に加える。
d) アスコルビン酸溶液(100 g/L) 19.2.2.1 f) による。
e) 硫酸ナトリウム JIS K 8987で規定するもの。
f)
1-ブタノール JIS K 8810で規定するもの。
g) シリカ標準液(SiO2:1 000 mg/L) 19.2.1.1 e) による。ただし,a) の水を用いて調製する。
h) シリカ標準液(SiO2:50 mg/L) シリカ標準液(SiO2:1 000 mg/L)25 mLを全量フラスコ500 mL
にとり,a) の水を標線まで加える。使用時に調製する。
i)
シリカ標準液(SiO2:2 mg/L) シリカ標準液(SiO2:50 mg/L)20 mLを全量フラスコ500 mLにと
り,a) の水を標線まで加える。使用時に調製する。
j)
シリカ標準液(SiO2:1 mg/L) シリカ標準液(SiO2:50 mg/L)10 mLを全量フラスコ500 mLにと
り,a) の水を標線まで加える。使用時に調製する。
19.2.3.2 器具及び装置
器具及び装置は,次による。
a) 分液漏斗 300 mL プラスチック製のもの。
b) 光度計 分光光度計又は光電光度計。
19.2.3.3 操作
操作は,次による。
a) 5.3によってろ過した試料の200 mLを分析漏斗にとる。イオン状シリカ濃度が100 µg/L以上の場合は,
試料の適量をとり,水を加えて200 mLとする。
b) 硫酸(2.5 mol/L)-モリブデン酸アンモニウム(188 g/L)混合溶液4 mLを加えて振り混ぜた後,液温
を約25 ℃に保ち約20分間放置する。
c) 硫酸(2+1)25 mLを加えて振り混ぜた後,直ちにアスコルビン酸溶液(100 g/L)2 mLを加えて振り
80
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
混ぜ,10分間放置する。
d) 1-ブタノール25 mLを加え,約2分間振り混ぜてモリブデン青を抽出する。
e) 静置後,1-ブタノール層を共栓試験管10 mLに入れ,硫酸ナトリウムを加えて脱水する。
f)
これを吸収セル20 mmに移し,1-ブタノールを対照液として波長800 nmの吸光度を測定する。試料
中のシリカの濃度がSiO2:10 µg/L以上の場合には,吸収セル10 mmを用いてもよい。ただし,空試
験及び検量線の作成における吸光度の測定にも吸収セル10 mmを用いる。
g) 次の操作によって,加えた硫酸(2.5 mol/L)-モリブデン酸アンモニウム(188 g/L)混合溶液に基づく
空試験値を求め,試料について得た吸光度を補正する。
分液漏斗(A)及び(B)それぞれに水200 mLをとり,硫酸(2.5 mol/L)-モリブデン酸アンモニウ
ム(188 g/L)混合溶液を(A)には4 mL,(B)には8 mLを加えて振り混ぜる。液温を約25 ℃に保
って20分間放置する。次に,c)〜f) の操作を行って(A),(B)それぞれについての吸光度a,bを測
定する。次の式によって硫酸(2.5 mol/L)-モリブデン酸アンモニウム(188 g/L)混合溶液による空試
験値cを算出する。
c=b−a
h) あらかじめ,次によって作成したシリカの検量線から,試料中のシリカの濃度(SiO2:µg/L)を算出
する。
1) 20 mmセルの場合,シリカ標準液(SiO2:1 mg/L)0.5〜10 mLを分液漏斗に段階的にとり,10 mm
セルの場合,シリカ標準液(SiO2:2 mg/L)1〜10 mLを分液漏斗に段階的にとり,水を加えて200 mL
とした後,b)〜f) の操作を行う。別に,この操作に用いた水200 mLをとり,b)〜f) の操作を行っ
てシリカ標準液について得た吸光度を補正し,シリカの濃度(SiO2:µg/L)と吸光度との関係線を
作成する。
19.2.4
流れ分析法
試料を5.3でろ過した後,試料中のシリカイオンを19.2.2と同様な原理で発色させる流れ分析法によっ
て定量する。
定量範囲:SiO2:0.02〜2 mg/L,繰返し精度:10 %以下(装置,測定条件によって異なる。)
19.2.4.1 モリブデン青発色FIA法
19.2.4.1.1 試薬
試薬は,次による。調製した溶液は,必要に応じて脱気を行う。
a) 水 JIS K 0557で規定するA3又はA4の水。
b) 硫酸(0.15 mol/L)-モリブデン酸アンモニウム(11.3 g/L)混合溶液 JIS K 8951で規定する硫酸4 mL
を水約150 mLに冷却しながら混合する。これにJIS K 8905で規定する七モリブデン酸六アンモニウ
ム四水和物6 gを水約250 mLに溶かした溶液を加え,全量フラスコ500 mLに移し入れ,水を標線ま
で加える。調製後,直ちにプラスチック容器で保存する。
c) しゅう酸溶液 JIS K 8519で規定するしゅう酸二水和物4.8 gを水に溶かして500 mLとする。
d) アスコルビン酸溶液 JIS K 9502で規定するL(+)-アスコルビン酸6 gを水に溶かして500 mLとする。
この溶液は,使用前に調製する。
e) キャリヤ液 水を用いる。
f)
シリカ標準液(SiO2:10 mg/L) 19.2.2.1 g) による。
19.2.4.1.2 器具及び装置
装置の基本構成は,次による(図22参照)。
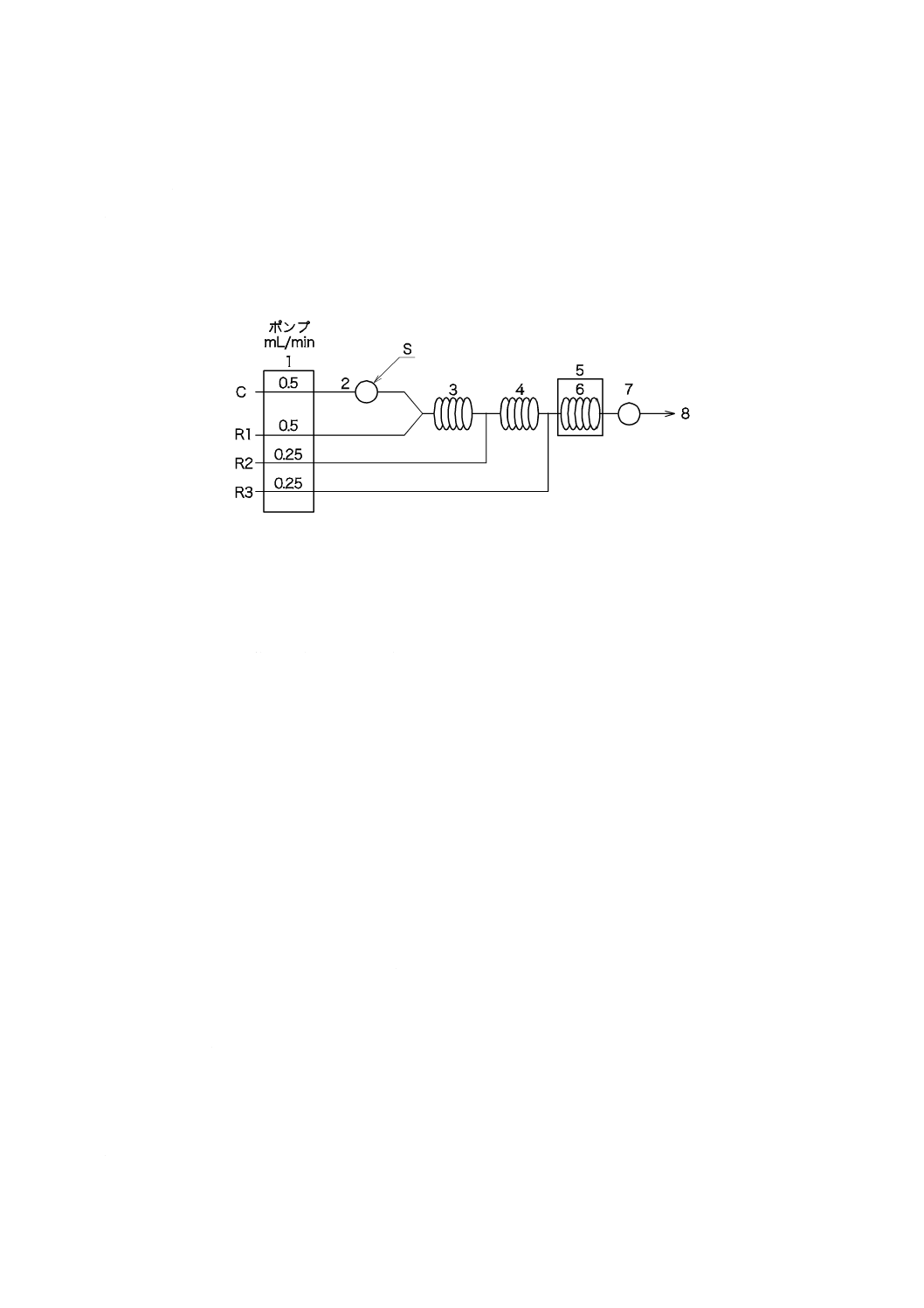
81
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) 送液部 15.7.1.2 a) による。
b) 試料導入部 15.7.1.2 b) による。
c) 反応部 15.7.1.2 c) 及び一定の温度に加熱保持できる恒温槽から構成する。
d) 検出部 波長815 nm付近での測定が可能な吸光光度検出器を用いる。
e) 記録部 15.7.1.2 e) による。
f)
恒温槽 80 ℃に加熱できるもの。
C: キャリヤ液
R1: 硫酸(0.15 mol/L)-モリブデン酸アンモニウム(11.3 g/L)混合溶液
R2: しゅう酸溶液
R3: アスコルビン酸溶液
S: 試料
1: ポンプ
2: 試料導入器(注入量500 μL)
3: 反応コイル(内径0.5 mm,長さ10 m)
4: 反応コイル(内径0.5 mm,長さ5 m)
5: 恒温槽(80 ℃)
6: 反応コイル(内径0.5 mm,長さ10 m)
7: 検出器(波長815 nm)
8: 廃液
図22−モリブデン青発色FIA法のシステム例
19.2.4.1.3 準備操作
準備操作は,15.7.1.3による。ただし,感度の調節はシリカによる。
19.2.4.1.4 操作
操作は,15.7.1.4による。ただし,検量線は,19.2.4.1.1 f) を水で希釈して調製した検量線用シリカ標準
液を用いて作成し,試料中のシリカ濃度の算出には,これを用いる。
19.2.4.1.5 留意事項
留意事項は,15.7.1.5による。
19.2.4.2 モリブデン青発色CFA法
19.2.4.2.1 試薬
試薬は,次による。調製した溶液は,必要に応じて脱気を行う。
a) 水 19.2.4.1.1 a) による。
b) ドデシル硫酸ナトリウム溶液 ドデシル硫酸ナトリウム15 gを87 mLの水に溶解させる。
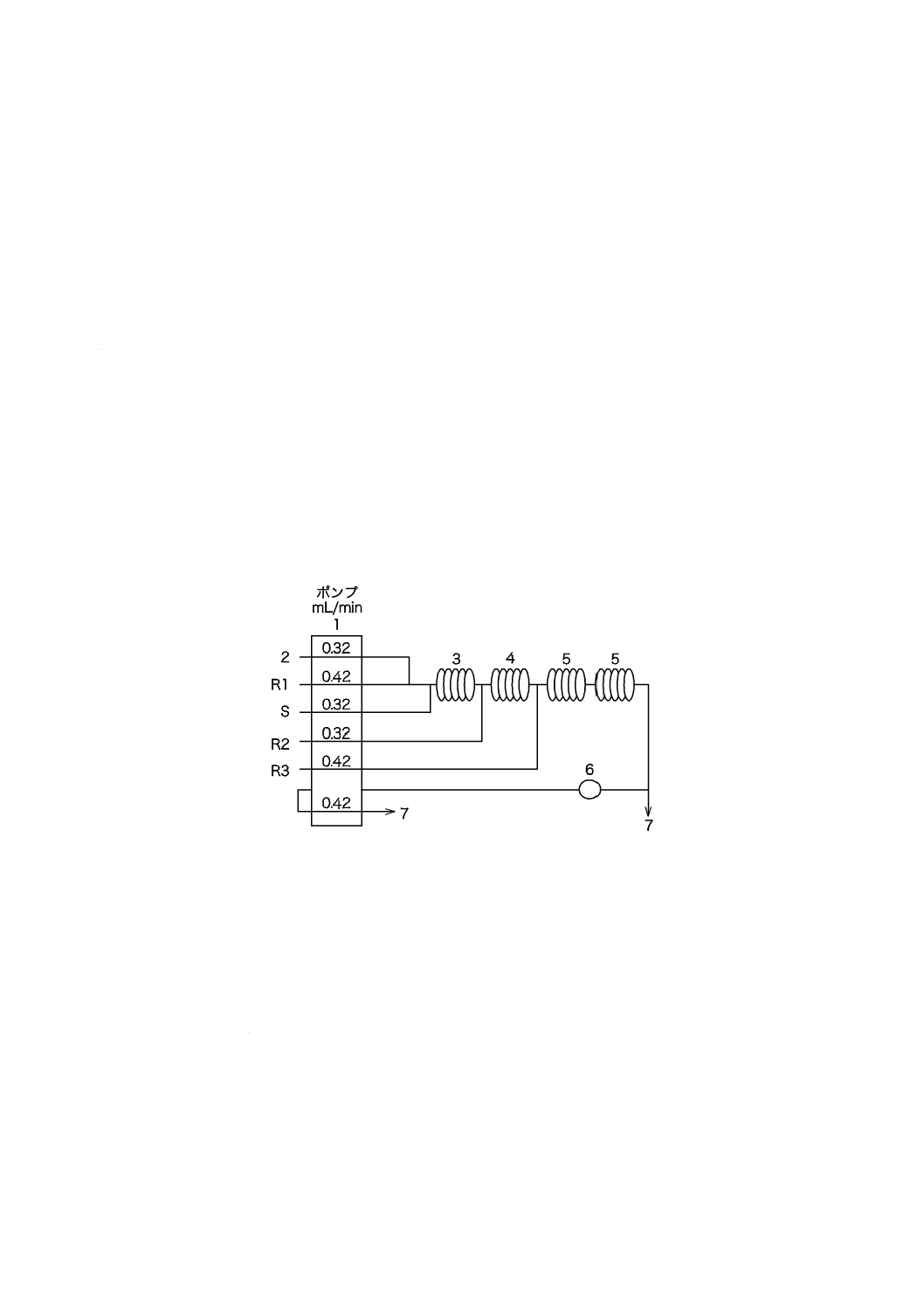
82
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
c) モリブデン酸アンモニウム溶液 JIS K 8951で規定する硫酸2.7 mLを水約800 mLに冷却しながら混
合する。これにJIS K 8905で規定する七モリブデン酸六アンモニウ四水和物10 gを加えて溶解し,全
量フラスコ1 000 mLに移し入れ,水を標線まで加える。これにドデシル硫酸ナトリウム溶液20 mL
を加えて混合する。調製後,直ちにプラスチック容器で保存する。
d) しゅう酸溶液 JIS K 8519で規定するしゅう酸二水和物30 gを水約800 mLに溶解し,全量フラスコ
1 000 mLに移し入れ,水を標線まで加える。調製後,直ちにプラスチック容器で保存する。
e) アスコルビン酸溶液 JIS K 9502で規定するL(+)-アスコルビン酸35 gを水約800 mLに溶解し,全
量フラスコ1 000 mLに移し入れ,水を標線まで加える。調製後,直ちにプラスチック容器で保存する。
f)
シリカ標準液(SiO2:10 mg/L) 19.2.2.1 g) による。
19.2.4.2.2 器具及び装置
装置の基本構成は,次による(図23参照)。
a) 送液部 15.7.2.2 a) による。
b) 試料導入部 15.7.2.2 b) による。
c) 反応部 15.7.2.2 c) による。
d) 検出部 波長800 nm付近での測定が可能な吸光光度検出器を用いる。
e) 記録部 15.7.2.2 e) による。
R1: モリブデン酸アンモニウム溶液
R2: しゅう酸溶液
R3: アスコルビン酸溶液
S: 試料
1: ポンプ
2: セグメントガス(空気)
3: 反応コイル(内径2 mm,長さ60 cm)
4: 反応コイル(内径2 mm,長さ30 cm)
5: 反応コイル(内径2 mm,長さ120 cm)
6: 検出器(波長800 nm)
7: 廃液
図23−モリブデン青発色CFA法のシステム例
19.2.4.2.3 準備操作
準備操作は,15.7.2.3による。ただし,感度の調節はシリカによる。
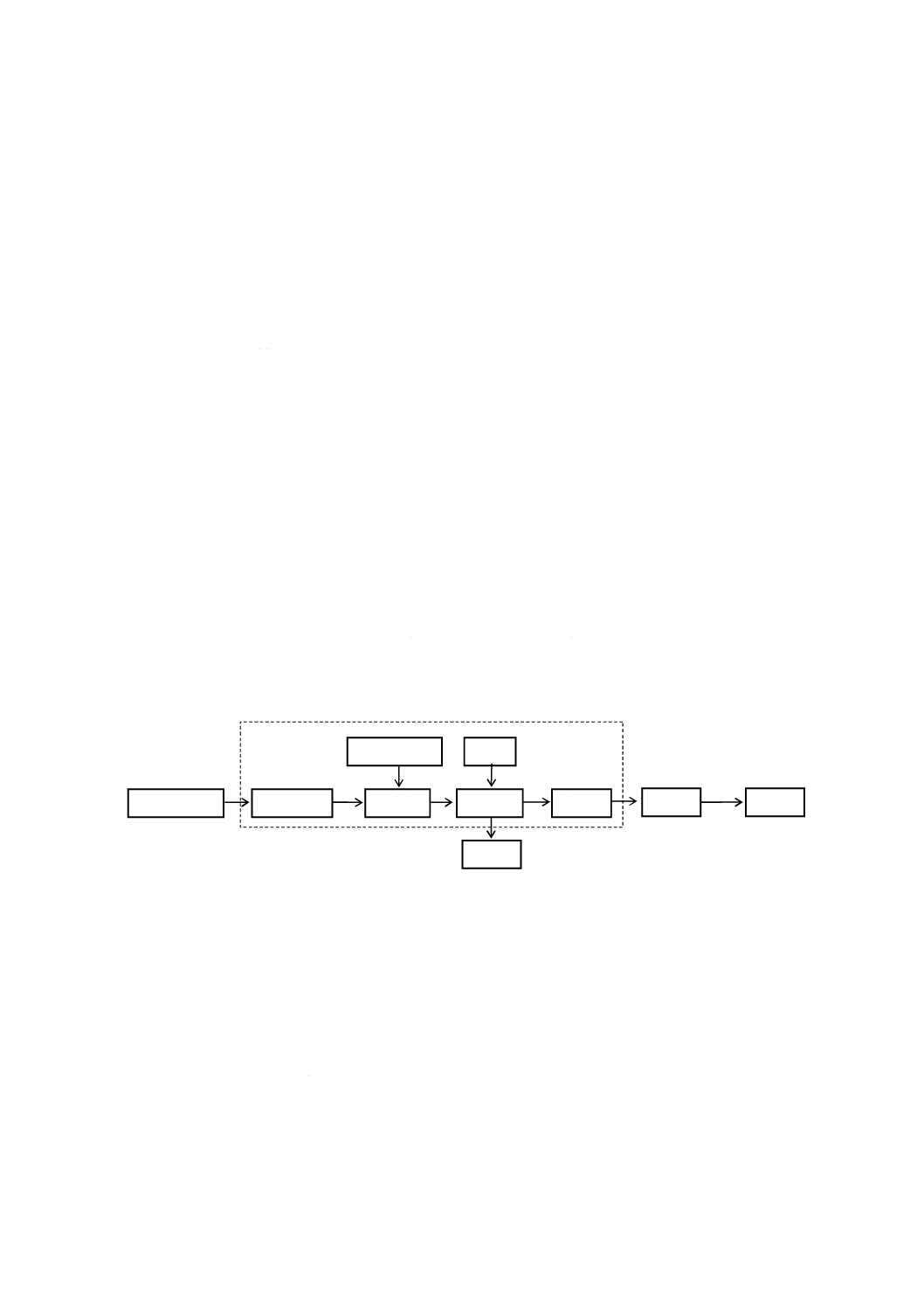
83
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
19.2.4.2.4 操作
操作は,15.7.2.4による。ただし,検量線は,19.2.4.2.1 f) を水で希釈して調製した検量線用シリカ標準
液を用いて作成し,試料中のシリカ濃度の算出には,これを用いる。
19.2.4.2.5 留意事項
留意事項は,15.7.2.5による。
19.2.5
シリカ(SiO2)プロセス用分析装置(モリブデン青吸光光度法)による測定方法
給水及びボイラ水中のシリカイオンを19.2.2と同様な原理で発色させ,プロセス用分析装置の光度計で
吸光度を測定してシリカを定量する。
測定範囲:SiO2 0〜500 µg/L,0〜2 mg/L(試験目的によって低濃度,高濃度のレンジ切替えが可能な方
式とする。),繰返し精度:最大目盛値の±5 %
19.2.5.1 試薬
試薬は,次による。
a) 水 19.2.1.1 a) による。
b) 発色試薬 それぞれのプロセス用分析装置の取扱説明書に従って調製する。
c) シリカ標準液(SiO2:100 mg/L) 19.2.1.1 f) による。
d) 校正液 シリカ標準液(SiO2:100 mg/L)を用いて,測定レンジの25,50,75 %に相当するシリカ濃
度の校正液を調製する。
19.2.5.2 器具及び装置
装置の構成は,次による。
a) 構成 プロセス用分析装置は,検出部,指示部及び記録部で構成する。構成例を図24に示す。
図24−プロセス用分析装置の構成例
19.2.5.3 測定
測定は,次による。
19.2.5.3.1 測定準備
プロセス用分析装置の各部を点検し,特に,水漏れのないことを確認した後,所定の手順に従って電源
を入れ,30分間以上装置を安定させる。
a) 発色試薬を試薬タンクに補給する。
19.2.5.3.2 校正
プロセス用分析装置が安定状態に達した後,次の操作を行う。
a) ゼロ校正 測定開始時及び試薬を調製し直したときなどに,水を用いてゼロ校正を行うか,又は電気
試薬供給部
光源
試料採取装置
温度調節器
反応槽
検出器
排出
指示部
記録部
試料セル
検出部
84
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
的にゼロ校正を行う。
b) スパン校正 測定開始時及び連続測定の場合は,2週間に1回程度の割合で,測定レンジの25,50,
75 %に相当する校正液を用いてスパン校正を行う。校正液の代わりに校正板を光源と試料セルの間に
挿入して,校正板のシリカの相当濃度を求めておき,その後の日常のスパン校正には,これを用いて
もよい。
19.2.5.3.3 測定
校正が終了したら試料を設定流量で流し,指示値が安定した後,連続測定を行う。
19.2.5.4 点検・整備
点検・整備は,定期的に行うこととし,次による。
a) 試料の流量
b) 試薬の補給
c) 試薬の添加量の確認
d) 試料のろ過器の交換
e) 記録紙及びインクの補給又は交換
19.3
溶存及びコロイド状シリカ
溶存及びコロイド状シリカは,試料をろ過したとき,ろ液に含まれるシリカをいう。溶存及びコロイド
状シリカの定量には,試料を前処理してシリカをイオン状とした後,モリブデン黄吸光光度法,モリブデ
ン青吸光光度法又は流れ分析法を適用する。
19.3.1
試料の前処理
試料をろ過し,炭酸水素ナトリウムを加え,煮沸してコロイド状シリカをイオン状とする。
19.3.1.1 試薬
試薬は,次による。
a) 水 19.2.1.1 a) による。
b) 塩酸(1+1) 19.2.1.1 b) による。
c) 炭酸水素ナトリウム JIS K 8622で規定するもので,SiO2の含量0.002 %以下のもの。
19.3.1.2 器具及び装置
器具及び装置は,次による。
a) 四ふっ化エチレン樹脂製ビーカ 200 mL
b) 白金皿
c) 光度計 分光光度計又は光電光度計。
19.3.1.3 操作
操作は,次による。
a) 5.3によってろ過した試料100 mLを四ふっ化エチレン樹脂製ビーカ200 mL(又は白金皿)にとり,
水を加えて50〜100 mLとする。
b) 試料100 mLについて炭酸水素ナトリウム0.20 gを加え,沸騰水浴中に入れ約20分間加熱する。
c) 放冷後,塩酸(1+1)で中和し,pHを約5とした後,全量フラスコ100 mLに移し入れ,水を標線ま
で加える。
d) 水100 mLを四ふっ化エチレン樹脂製ビーカ200 mL(又は白金皿)にとり,b)〜c) の操作を行って空
試験用試料とする。
19.3.2
モリブデン黄吸光光度法
85
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
19.3.1で処理した試料に,19.2.1のモリブデン黄吸光光度法を適用し,試料水中の溶存及びコロイド状シ
リカ濃度(SiO2:mg/L)を求める。試薬,装置,操作は19.2.1による。
19.3.3
モリブデン青吸光光度法
19.3.1で処理した試料に,19.2.2のモリブデン青吸光光度法を適用し,試料水中の溶存及びコロイド状シ
リカ濃度(SiO2:mg/L)を求める。試薬,装置,操作は19.2.2による。
19.3.4
流れ分析法
19.3.1で処理した試料に,19.2.4の流れ分析法を適用し,試料水中の溶存及びコロイド状シリカ濃度
(SiO2:mg/L)を求める。試薬,装置,操作は19.2.4による。
19.4
全シリカ
全シリカは水中に含まれる全てのシリカをいう。全シリカの定量には,試料を前処理してシリカをイオ
ン状とした後,モリブデン黄吸光光度法,モリブデン青吸光光度法又は流れ分析法を適用するか,前処理
が不要な重量法を適用する。
19.4.1
吸光光度法の試料の前処理(炭酸ナトリウムによる融解)
試料に炭酸ナトリウムを加えて蒸発乾固し,融解して全てのシリカをイオン状にする。
19.4.1.1 試薬
試薬は,次による。
a) 水 19.2.1.1 a) による。
b) 塩酸(1+1) 19.2.1.1 b) による。
c) 炭酸ナトリウム JIS K 8005で規定するもので,SiO2の含量0.001 %以下のもの。
19.4.1.2 器具及び装置
器具及び装置は,次による。
a) 四ふっ化エチレン樹脂製ビーカ 200 mL(又は白金皿)。
b) 白金るつぼ
c) 光度計 分光光度計又は光電光度計。
19.4.1.3 操作
操作は,次による。
a) 試料100 mLを四ふっ化エチレン樹脂製ビーカ200 mL(又は白金皿)にとり,炭酸ナトリウム0.20 g
を加え,沸騰水浴中に入れ,加熱蒸発して約5 mLに濃縮する。
b) 内容物を少量の水で白金るつぼに移し,再び蒸発して乾固する。
c) 静かに加熱して有機物を炭化した後,灰化し,強熱して融解し,放冷する。
d) 水を加え,加熱して融成物を溶かし,放冷後,ビーカに移し,塩酸(1+1)で中和してpHを約5と
する。
e) 濁りがある場合には,ろ過して水で洗浄し,ろ液と洗液とを全量フラスコ100 mLに移し入れ,水を
標線まで加える。
19.4.2
モリブデン黄吸光光度法
19.4.1で処理した試料に,19.2.1のモリブデン黄吸光光度法を適用し,試料水中の全シリカ濃度(SiO2:
mg/L)を求める。試薬,装置,操作は19.2.1による。
19.4.3
モリブデン青吸光光度法
19.4.1で処理した試料に,19.2.2のモリブデン青吸光光度法を適用し,試料水中の全シリカ濃度(SiO2:
mg/L)を求める。試薬,装置,操作は19.2.2による。
86
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
19.4.4
流れ分析法
19.4.1で処理した試料に,19.2.4の流れ分析法を適用し,試料水中の全シリカ濃度(SiO2:µg/L)を求め
る。試薬,装置,操作は19.2.4による。
19.4.5
重量法
試料に塩酸と過塩素酸とを加えて加熱し,過塩素酸の白煙を発生させ,シリカを脱水して不溶性にする。
水を加えて塩類を溶かした後,シリカをろ別し,加熱して恒量とした後,硫酸とふっ化水素酸でシリカを
揮散させ,その減量からシリカを定量する。
定量範囲:SiO2:50 mg/L以上,繰返し精度:3〜10 %
19.4.5.1 試薬
試薬は,次による。
a) 水 19.2.1.1 a) による。
b) 塩酸(1+1) 19.2.1.1 b) による。
c) 塩酸(1+50) JIS K 8180で規定する塩酸を用いて調製する。
d) 硫酸(1+2) 水2容をビーカにとり,これを冷却し,かき混ぜながら,JIS K 8951で規定する硫酸
1容を徐々に加える。
e) 過塩素酸 JIS K 8223で規定するもの。
f)
ふっ化水素酸 JIS K 8819で規定するもの。
g) 硝酸 JIS K 8541で規定するもの。
19.4.5.2 器具及び装置
器具及び装置は,次による。
a) 白金るつぼ
19.4.5.3 操作
操作は,次による。
a) 試料の適量(SiO2 5 mg以上を含む。)をビーカにとり,塩酸(1+1)10 mLと過塩素酸15 mLとを加
える。ただし,試料中に有機物が多量に含まれている場合には,試料に硝酸を加えて酸性とした後,
加熱し,蒸発して液量が少量となったとき,硝酸10〜20 mLを加えて加熱する。放冷後,過塩素酸15
mLを加える。
b) 加熱蒸発し,過塩素酸の濃い白煙が発生し始めたら時計皿でビーカを覆い,引き続き約15分間加熱し,
放冷する。
c) 温水100 mLを加えて可溶性塩類を溶かし,ろ紙5種Bでろ別し,残留物及びろ紙を温塩酸(1+50)
で数回,次に,温水で数回洗浄する。
d) 残留物はろ紙とともに約1 000 ℃で恒量とした白金るつぼに入れ,乾燥後,徐々に加熱してろ紙を炭
化した後,灰化し,約1 000 ℃で加熱して恒量とする。
e) 硫酸(1+2)数滴を加えて残留物を湿し,ふっ化水素酸5 mLを加え,注意して加熱してシリカを揮
散させ,引き続き加熱乾燥後,約1 000 ℃で加熱して恒量とする。
f)
空試験として,あらかじめ約1 000 ℃で恒量とした白金るつぼに硫酸(1+2)数滴と,ふっ化水素酸
5 mLとを加えてe) の操作を行ってふっ化水素酸の強熱残分を求め,ふっ化水素酸の空試験値とする。
g) 次の式によって試料中のシリカの濃度(SiO2:mg/L)を算出する。
(
)
[
]
V
W
W
W
S
000
1
3
2
1
×
−
−
=
87
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ここに,
S: 全シリカ(SiO2:mg/L)
W1: d) の操作で得た残留物の質量(mg)
W2: e) の操作を行った後の残留物の質量(mg)
W3: ふっ化水素酸の空試験値(mg)
V: 試料(mL)
警告 過塩素酸を用いる加熱分解操作は,試料の種類によっては爆発の危険性があるため,次のこ
とに注意する。
i)
酸化されやすい有機物は,過塩素酸を加える前に,b) の操作によって十分に分解してお
く。
ii) 過塩素酸の添加は,必ず濃縮液を放冷した後に行う。
iii) 必ず過塩素酸と硝酸を共存させた状態で加熱分解を行う。
iv) 濃縮液を乾固させない。
警告 ふっ化水素酸は毒物であり取扱いに注意し,法令に従い安全及び健康に対する適切な措置を
取らなければならない。また,廃液の処分にも注意する。
20
ヒドラジン(N2H4)[ヒドラジニウムイオン(N2H5+)]
20.1
一般事項
ヒドラジンの定量には,p-ジメチルアミノベンズアルデヒド吸光光度法,よう素滴定法,流れ分析法又
は,プロセス用分析装置による測定方法を適用する。
ヒドラジンは,アルカリ性で不安定であるので,あらかじめ塩酸を添加した試料容器に試料を採取し,
できるだけ早く試験する。
20.2
p-ジメチルアミノベンズアルデヒド吸光光度法
試料を塩酸酸性にしてp-ジメチルアミノベンズアルデヒドを加えて生じる黄色の化合物の吸光度を測定
してヒドラジニウムイオンを定量する。
なお,ヒドラジニウムイオンN2H5+:1 mgは,ヒドラジンN2H4:0.969 5 mgに相当する。
定量範囲: 10 mmセル使用の場合N2H5+:20〜500 µg/L
20 mmセル使用の場合N2H5+:10〜240 µg/L
繰返し精度:3〜10 %
20.2.1
試薬
試薬は,次による。
a) 水 JIS K 0557で規定するA3の水。
b) 塩酸(1+9) JIS K 8180で規定する塩酸を用いて調製する。
c) 塩酸(1+99) JIS K 8180で規定する塩酸を用いて調製する。
d) p-ジメチルアミノベンズアルデヒド溶液 JIS K 8496で規定するp-ジメチルアミノベンズアルデヒド
4.0 gをJIS K 8891で規定するメタノール[又はJIS K 8102で規定するエタノール(95)]200 mLと
JIS K 8180で規定する塩酸20 mLとを混ぜた溶液中に溶かして着色ガラス瓶に保存する。
e) ヒドラジニウムイオン標準液(N2H5+:100 mg/L) 塩化ヒドラジニウム0.318 gをとり,JIS K 8180
で規定する塩酸10 mLと水約20 mLとを混ぜた溶液中に溶かし,全量フラスコ1 000 mLに移し入れ,
水を標線まで加える。使用時に調製する。
f)
ヒドラジニウムイオン標準液(N2H5+:1 mg/L) ヒドラジニウムイオン標準液(N2H5+:100 mg/L)
10 mLを全量フラスコ1 000 mLにとり,JIS K 8180で規定する塩酸10 mLを加え,水を標線まで加え
88
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
る。使用時に調製する。
20.2.2
器具及び装置
器具及び装置は,次による。
a) 光度計 分光光度計又は光電光度計。
b) メスシリンダ(有栓形) 15.2.2 b) による。
20.2.3
試料採取
試料採取は,次による。
a) 試料採取配管の先端に軟質塩化ビニル管(又はポリエチレン管)を接続し,試料の流量が200〜300
mL/minの割合で流れるように調節し,試料が完全に入れ替わるまで十分に流す。
b) あらかじめ,塩酸(1+9)10 mLを入れたメスシリンダ(有栓形)100 mLの塩酸溶液中に試料採取用
配管の先端(軟質塩化ビニル管の先端の部分)を差し込み,液量が100 mLになるまで採取する。
20.2.4
操作
操作は,次による。
a) 5.3によってろ過した試料50 mLをメスシリンダ(有栓形)50 mLにとる。ヒドラジウムイオン濃度
が500 µg /L以上の場合は,試料の適量をとり,塩酸(1+99)を加えて50 mLとする。
b) p-ジメチルアミノベンズアルデヒド溶液10 mLを加えてよく振り混ぜ,約10分間放置する。
c) これを吸収セルに移し,別に,塩酸(1+99)50 mLについてb) の操作を行った空試験の溶液を対照
液として波長458 nm付近の吸光度を測定する。
d) あらかじめ,次によって作成したヒドラジニウムイオンの検量線から,ヒドラジニウムイオンの濃度
(N2H5+:µg/L)を算出する。
1) セル長が10 mmの場合,ヒドラジニウムイオン標準液(N2H5+:1 mg/L)1〜25 mL,セル長が20 mm
の場合,ヒドラジニウムイオン標準液(N2H5+:1 mg/L)0.5〜12 mLをメスシリンダ(有栓形)50 mL
に段階的にとり,塩酸(1+99)を50 mLの標線まで加え,b) 及びc) の操作を行ってヒドラジニ
ウムイオン濃度(N2H5+:µg/L)と吸光度との関係線を作成する。
20.3
よう素滴定法
試料に炭酸水素ナトリウムを加えて弱アルカリ性とし,よう素溶液で滴定してヒドラジニウムイオンを
定量する。
定量範囲:N2H5+:4 mg/L以上
20.3.1
試薬
試薬は,次による。
a) 水 20.2.1 a) による。
b) 炭酸水素ナトリウム JIS K 8622で規定するもの。
c) でんぷん溶液(10 g/L) JIS K 8659で規定するでんぷん(溶性)1 gを水約10 mLに混ぜ,熱水100
mL中にかき混ぜながら加え,約1分間煮沸した後,放冷する。使用時に調製する。
d) 50 mmol/Lチオ硫酸ナトリウム溶液 JIS K 8637で規定するチオ硫酸ナトリウム五水和物12.5 gとJIS
K 8625で規定する炭酸ナトリウム0.2 gとを水に溶かし,水を加えて1 Lとする。2日間放置した後,
標定する。この溶液は使用時に,次によって標定して用いる。
1) JIS K 8005で規定する容量分析用標準物質のよう素酸カリウムを130 ℃で約2時間加熱し,デシケ
ータ中で放冷する。KIO3 100 %に対してその0.357 gをとり,少量の水に溶かし,全量フラスコ200
mLに移し入れ,水を標線まで加える。この20 mLを共栓三角フラスコ300 mLにとり,JIS K 8913
89
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
で規定するよう化カリウム2 g及び硫酸(1+5)5 mLを加え,直ちに栓をして静かに振り混ぜ,暗
所に約5分間放置する。これに水100 mLを加え,遊離したよう素をこのチオ硫酸ナトリウム溶液
で滴定する。溶液の黄色が薄くなってから,指示薬としてでんぷん溶液(10 g/L)1 mLを加え,生
じたよう素でんぷんの青い色が消えるまで滴定する。
別に,水について同一条件で空試験を行って補正したmL数から,次の式によって50 mmol/Lチ
オ硫酸ナトリウム溶液のファクタ( f )を算出する。
783
001
.0
1
200
20
100
×
×
×
×
=
x
b
a
f
ここに,
a: よう素酸カリウムの量(g)
b: よう素酸カリウムの純度(%)
x: 滴定に要した50 mmol/Lチオ硫酸ナトリウム溶液(補正
した値)(mL)
0.001 783: 50 mmol/Lチオ硫酸ナトリウム溶液1 mLのよう素酸カ
リウム相当量(g)
e) 25 mmol/Lよう素溶液 JIS K 8913で規定するよう化カリウム20 gを少量の水に溶かし,これにJIS K
8920で規定するよう素6.4 gを加えて溶かし,水を加えて1 Lとする。
標定 このよう素溶液20 mLを三角フラスコ300 mLにとり,水を加えて液量を約100 mLとし,50
mmol/Lチオ硫酸ナトリウム溶液で滴定し,溶液の色が薄くなったら,指示薬としてでんぷん
溶液(10 g/L)1 mLを加えて振り混ぜ,更によう素でんぷんの青い色が消えるまで滴定する。
次の式によって25 mmol/Lよう素溶液のファクタ( f1)を算出する。この標定は使用時に行
う。
20
1
f
a
f
×
=
ここに,
a: 滴定に要した50 mmol/Lチオ硫酸ナトリウム溶液(mL)
f: 50 mmol/Lチオ硫酸ナトリウム溶液のファクタ
20.3.2
試料採取
試料採取は,次による。
a) 20.2.3 a) に準じて試料を採取する。
20.3.3
操作
操作は,次による。
a) 試料の適量(N2H5+:4 mg/L以上を含む。)を三角フラスコ300 mLにとり,水を加えて100 mLとす
る。
b) 炭酸水素ナトリウム約2 g及びでんぷん溶液(10 g/L)1 mLを加えて振り混ぜる。
c) 25 mmol/Lよう素溶液で滴定し,よう素でんぷんの青い色が認められる(約30秒間)ときを終点とす
る。
d) 空試験として水100 mLを三角フラスコ300 mLにとり,b) 及びc) の操作を行う。
e) 次の式によって試料中のヒドラジニウムイオンの濃度(N2H5+:mg/L)を算出する。
(
)
2
413
.0
000
1
1
×
×
×
−
=
V
f
b
a
N
ここに,
N: ヒドラジニウムイオン(N2H5+:mg/L)
a: 滴定に要した25 mmol/Lよう素溶液(mL)
b: 空試験の滴定に要した25 mmol/Lよう素溶液(mL)
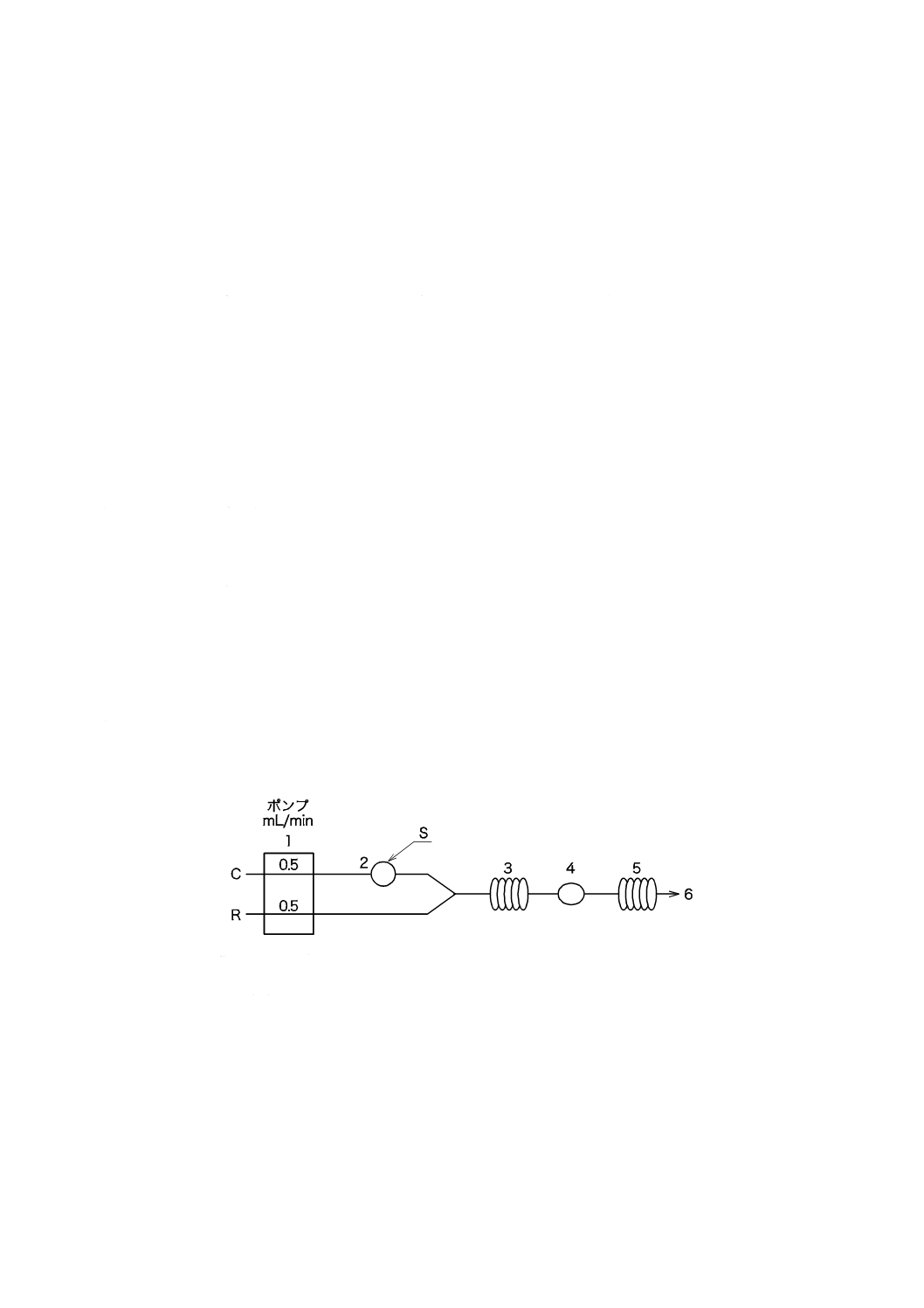
90
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
f1: 25 mmol/Lよう素溶液のファクタ
V: 試料(mL)
0.413 2: 25 mmol/Lよう素溶液1 mLのヒドラジニウムイオン相
当量(mg)
20.4
流れ分析法
試料を5.3によってろ過した後,試料中のヒドラジニウムイオンを20.2と同様な原理で発色させる流れ
分析法によって定量する。
定量範囲:N2H5+:10〜500 µg/L,繰返し精度:10 %以下(装置,測定条件によって異なる。)
20.4.1 p-ジメチルアミノベンズアルデヒド発色FIA法
20.4.1.1 試薬
試薬は,次による。調製した溶液は,必要に応じて脱気を行う。
a) 水 20.2.1 a) による。
b) p-ジメチルアミノベンズアルデヒド溶液 JIS K 8496で規定するp-ジメチルアミノベンズアルデヒド
4.0 gをJIS K 8102で規定するエタノール(95)200 mLとJIS K 8180で規定する塩酸20 mLとを混ぜ
た溶液中に溶かして着色ガラス瓶に保存する。
c) キャリヤ液 水を用いる。
d) 塩酸(1+99) 20.2.1 c) による。JIS K 8180で規定する塩酸を用いて調製する。
e) ヒドラジニウムイオン標準液(N2H5+:1 mg/L) 20.2.1 f) による。
20.4.1.2 器具及び装置
装置の基本構成は,次による(図25参照)。
a) 送液部 15.7.1.2 a) による。
b) 試料導入部 15.7.1.2 b) による。
c) 反応部 15.7.1.2 c) による。
d) 検出部 波長458 nm付近での測定が可能な吸光光度検出器を用いる。
e) 記録部 15.7.1.2 e) による。
C: キャリヤ液
R: p-ジメチルアミノベンズアルデヒド溶液
S: 試料
1: ポンプ
2: 試料導入器(注入量200 μL)
3: 反応コイル(内径0.5 mm,長さ10 m)
4: 検出器(波長458 nm)
5: 背圧コイル(内径0.5 mm,長さ5 m)
6: 廃液
図25−p-ジメチルアミノベンズアルデヒド発色FIA法のシステム例
91
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
20.4.1.3 準備操作
準備操作は,15.7.1.3による。ただし,感度の調節はヒドラジニウムイオンによる。
20.4.1.4 操作
操作は,15.7.1.4による。ただし,検量線は,20.4.1.1 e) を塩酸(1+99)で希釈して調製した検量線用
ヒドラジニウムイオン標準液を用いて作成し,試料中のヒドラジニウムイオン濃度の算出には,これを用
いる。
20.4.1.5 留意事項
留意事項は,15.7.1.5による。
20.4.2 p-ジメチルアミノベンズアルデヒド発色CFA法
20.4.2.1 試薬
試薬は,次による。調製した溶液は,必要に応じて脱気を行う。
a) 水 20.2.1 a) による。
b) ドデシル硫酸ナトリウム溶液 ドデシル硫酸ナトリウム15 gを87 mLの水に溶解させる。
c) 硫酸溶液 JIS K 8951で規定する硫酸2 mLを水約800 mLに冷却しながら混合する。これを全量フラ
スコ1 000 mLに移し入れ,水を標線まで加える。これにドデシル硫酸ナトリウム溶液20 mLを加え
て混合する。
d) p-ジメチルアミノベンズアルデヒド溶液 JIS K 8951で規定する硫酸50 mLを水約700 mLに冷却し
ながら混合する。これにJIS K 8496で規定するp-ジメチルアミノベンズアルデヒド5 gとJIS K 8891
で規定するメタノール100 mLとを加えて溶解する。これを全量フラスコ1 000 mLに移し入れ,水を
標線まで加える。
e) 塩酸(1+99) 20.2.1 c) による。JIS K 8180で規定する塩酸を用いて調製する。
f)
ヒドラジニウムイオン標準液(N2H5+:1 mg/L) 20.2.1 f) による。
20.4.2.2 器具及び装置
装置の基本構成は,次による(図26参照)。
a) 送液部 15.7.2.2 a) による。
b) 試料導入部 15.7.2.2 b) による。
c) 反応部 15.7.2.2 c) 及び一定の温度に加熱保持できる恒温槽から構成する。
d) 検出部 波長458 nm付近での測定が可能な吸光光度検出器を用いる。
e) 記録部 15.7.2.2 e) による。
f)
恒温槽 45 ℃に加熱できるもの。
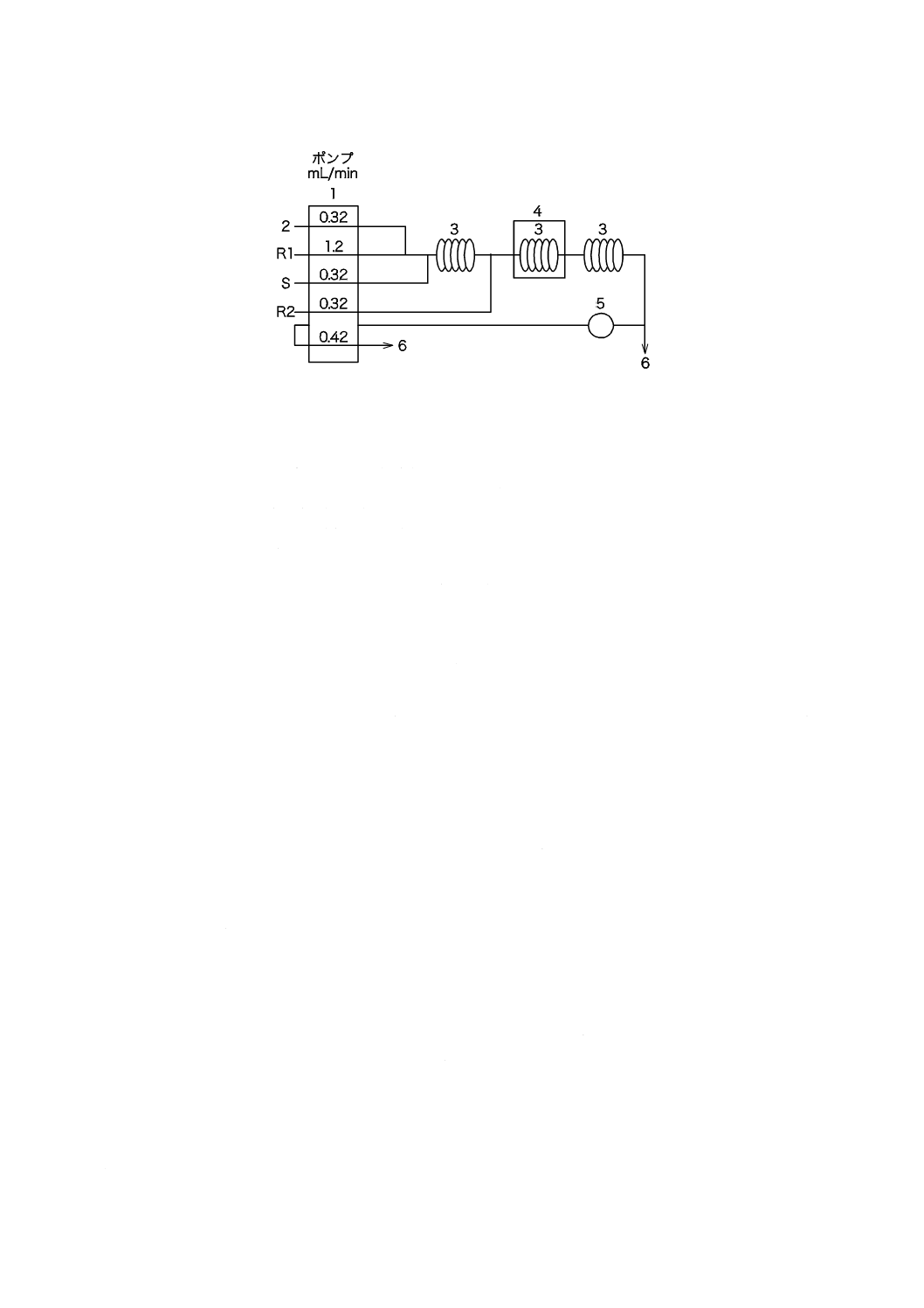
92
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
S: 試料
R1: 硫酸溶液
R2: p-ジメチルアミノベンズアルデヒド溶液
1: ポンプ
2: セグメントガス(空気)
3: 反応コイル(内径2 mm,長さ30 cm)
4: 恒温槽(45 ℃)
5: 検出器(波長458 nm)
6: 廃液
図26−p-ジメチルアミノベンズアルデヒド発色CFA法のシステム例
20.4.2.3 準備操作
準備操作は,15.7.2.3による。ただし,感度の調節はヒドラジニウムイオンによる。
20.4.2.4 操作
操作は,15.7.2.4による。ただし,検量線は,20.4.2.1 f) を塩酸(1+99)で希釈して調製した検量線用
ヒドラジニウムイオン標準液を用いて作成し,試料中のヒドラジニウムイオン濃度の算出には,これを用
いる。
20.4.2.5 留意事項
留意事項は,15.7.2.5による。
20.5
ヒドラジン(N2H4)[ヒドラジニウムイオン(N2H5+)]プロセス用分析装置(酸化還元電極)によ
る測定方法
給水中のヒドラジニウムイオンをプロセス用分析装置内電極によって連続的に測定する。
測定範囲:N2H5+ 0〜50 µg/L,0〜100 µg/L,0〜200 µg/L(試験目的によって濃度のレンジ切替えが可能
な方式とする。),繰返し精度:最大目盛値の±5 %
20.5.1
試薬
試薬は,次による。
a) ヒドラジニウムイオン標準液(N2H5+:100 mg/L) 20.2.1 e) による。
b) 校正液 ヒドラジニウムイオン標準液(N2H5+:100 mg/L)を用いて,測定レンジの25,50,75 %に
相当する濃度の校正液を調製する。校正液は使用時に調製する。
20.5.2
器具及び装置
装置の構成は,次による。
a) 構成 プロセス用分析装置は,検出部,指示部及び記録部で構成する。構成例を図27に示す。
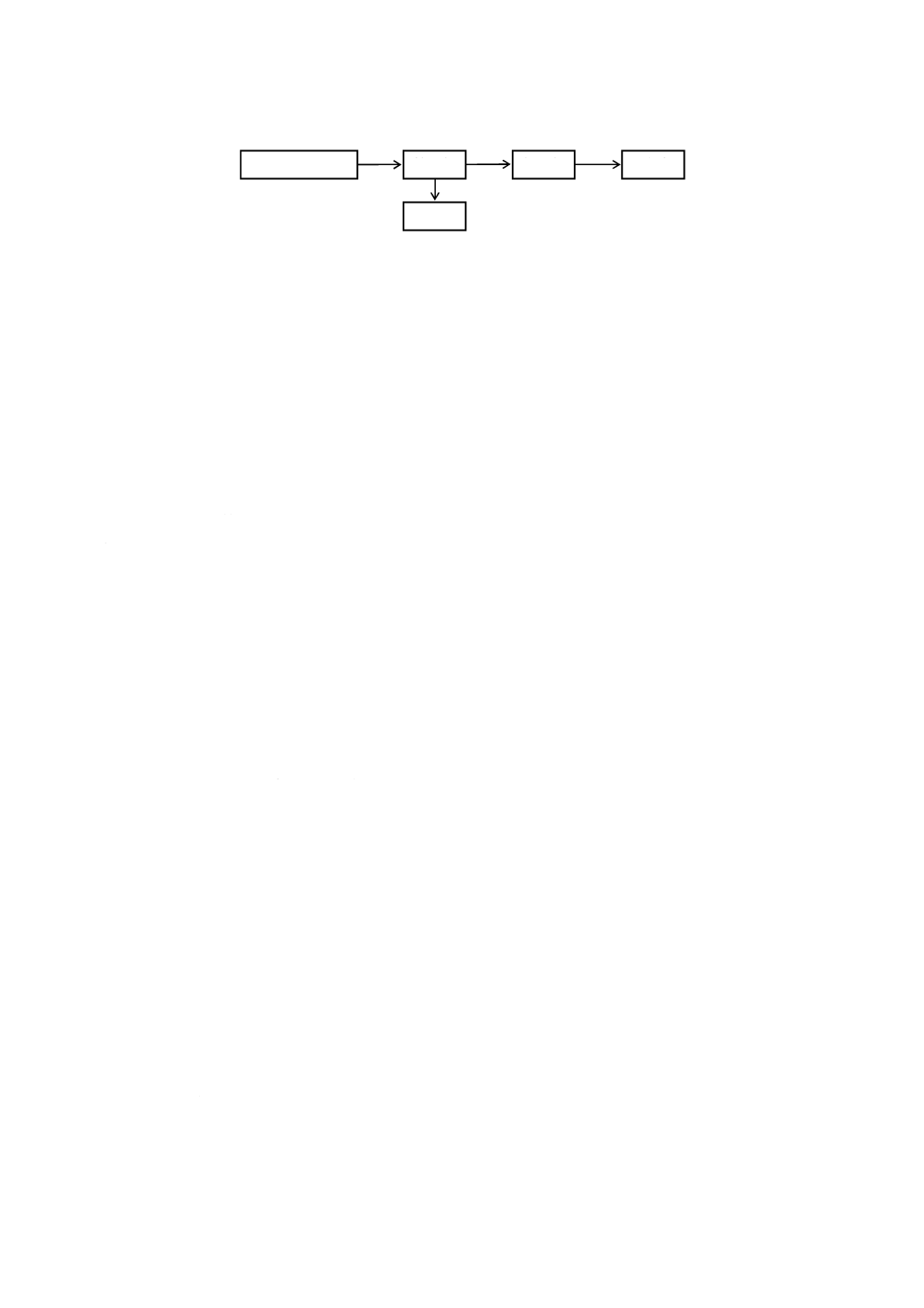
93
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図27−プロセス用分析装置の構成例
20.5.3
測定
測定は,次による。
20.5.3.1 測定準備
プロセス用分析装置の各部を点検し,特に,水漏れのないことを確認してから,所定の手順に従って電
源を入れ,30分間以上装置を安定させる。
20.5.3.2 校正
プロセス用分析装置が安定状態に達した後,次の操作を行う。
a) ゼロ校正 ヒドラジニウムイオン濃度がゼロとなるような電気的回路条件にすることによって,指示
値をゼロに調節する。
b) スパン校正 測定レンジの25,50,75 %に相当する校正液を用いてスパン校正を行う。スパン校正は,
2〜4週間に1回程度実施することが望ましい。
20.5.3.3 測定
校正が終了したら試料を設定流量で流し,指示値が安定した後,連続測定を行う。
20.5.4
点検・整備
点検・整備は,定期的に行うこととし,次による。
a) 試料の流量
b) 検出部(電極)の汚れ
c) 内筒液の補給
d) 記録紙及びインクの補給又は交換
21
ナトリウム(Na)
21.1
一般事項
ナトリウムの定量には,フレーム光度法,フレーム原子吸光法,電気加熱原子吸光法,ICP発光分光分
析法,ICP質量分析法,イオン電極法,イオンクロマトグラフ法又はプロセス用分析装置による測定法を
適用する。極低濃度域の測定については,電気加熱原子吸光法,ICP質量分析法又はプロセス用分析装置
による測定法を適用する。
21.2
フレーム光度法
試料をアセチレン-空気フレームの中に噴霧し,このときに生じる波長589.0 nmの輝線の強さを測定し
てナトリウムを定量する。
定量範囲:Na:30〜300 µg/L,0.3〜3 mg/L,3〜30 mg/L,繰返し精度:3〜10 %(装置及び測定条件に
よって異なる。)
21.2.1
試薬
試薬は,次による。
試料採取装置
検出部
指示部
記録部
排出水
94
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) 水 JIS K 0557で規定するA2又はA3の水。
b) ナトリウム標準液(Na:1 000 mg/L) 国家計量標準にトレーサブルなナトリウム標準液(Na:1 000
mg/L)を使用するか,又は,次による。JIS K 8005で規定する容量分析用標準物質の塩化ナトリウム
を600 ℃で約1時間加熱し,デシケータ中で放冷する。NaCl 100 %に対してその2.542 gをとり,少
量の水に溶かして全量フラスコ1 000 mLに移し入れ,水を標線まで加える。ポリエチレン瓶に保存す
る。
c) ナトリウム標準液(Na:3〜30 mg/L) ナトリウム標準液(Na:1 000 mg/L)を段階的にとり,これ
を水で薄めてNa:3〜30 mg/Lの標準液を調製する。
なお,低い濃度の測定用には,Na:30〜300 µg/L又はNa:0.3〜3 mg/Lの標準液を調製して用いる。
21.2.2
器具及び装置
器具及び装置は,次による。
a) フレーム光度計 炎光による分光測定が可能なもの。
21.2.3
操作
操作は,次による。
a) ナトリウム標準液(Na:30 mg/L)をフレーム光度計のフレーム中に噴霧し,波長589.0 nmの指示値
が100を示すように調節する。
b) 水を噴霧して指示値がゼロを示すように調節する。
c) ナトリウム標準液(Na:3〜30 mg/L)を,順次,噴霧し,ナトリウム(Na)の濃度と指示値との関係
線を作成し,検量線とする。
d) 試料(ナトリウムの濃度がNa:30 mg/L以上の場合は薄める。)を噴霧して指示値を読み取り,検量
線から試料中のナトリウムの濃度(mg/L)を求める。
21.2.4
留意事項
a) 試料に懸濁物が含まれる場合には,ろ過又は遠心分離によって懸濁物を除去する。
b) 試料がアルカリ性の場合には,硝酸(1+1)を滴加して弱酸性にする。硝酸(1+1)の滴加量が多い
場合には,測定したナトリウムの濃度(mg/L)を希釈割合に応じて補正する。
c) 干渉物質が含まれる場合には,その影響を無視できる濃度まで薄めて測定するか試料と同程度の干渉
物質を含むナトリウム標準液(Na:3〜30 mg/L)を調製し,検量線を作成する。
d) カリウム及びカルシウムが共存すると正の誤差を生じる。このような試料には,塩化セシウム溶液(25
g/L)[塩化セシウム25 gをJIS K 8180で規定する塩酸50 mL及び水450 mLに溶かし,水を加えて1 L
とする。この溶液1 Lは,セシウム(Cs)を約20 gを含む。]を試料40 mLに対して5 mLを加えるこ
とで,カリウム,カルシウムなどの影響を抑制できる。この操作を行った場合は,検量線作成時の操
作も塩化セシウム(25 g/L)を試料と同様に加えて行う。
e) リチウム,バリウム,遊離酸,りん酸塩,ほう酸塩,しゅう酸塩,シリカ,グルコース,ゼラチンな
どが共存すると負の誤差を生じる。
f)
マグネシウムと硫酸イオンとはほとんど干渉しない。
g) 多量のけい酸塩が共存する場合には,試料の適量を石英ガラス製ビーカ又は白金皿にとり,塩酸(1
+1)を加えて酸性とした後,蒸発乾固する。放冷後,塩酸(1+1)5滴及び少量の水を加え,加熱し
て溶かし,ろ過して水で一定量とする。
21.3
フレーム原子吸光法
試料をアセチレン-空気フレーム中に噴霧し,ナトリウムによる原子吸光を波長589.0 nmで測定して,
95
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ナトリウムを定量する。
定量範囲:Na:0.05〜4 mg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
21.3.1
試薬
試薬は,次による。
a) 水 21.2.1 a) による。
b) ナトリウム標準液(Na:100 mg/L) 国家計量標準にトレーサブルなナトリウム標準液(Na:100 mg/L)
を使用するか,又は,次による。21.2.1 b) のナトリウム標準液(Na:1 000 mg/L)10 mLを全量フラ
スコ100 mLにとり,水を標線まで加える。使用時に調製する。
c) ナトリウム標準液(Na:10 mg/L) ナトリウム標準液(Na:100 mg/L)20 mLを全量フラスコ200 mL
にとり,水を標線まで加える。使用時に調製する。
21.3.2
器具及び装置
器具及び装置は,次による。
a) フレーム原子吸光分析装置 JIS K 0121で規定するフレーム原子吸光分析装置で,測定対象元素用の
光源を備え,かつ,バックグラウンド補正が可能なもの。
21.3.3
操作
操作は,次による。
a) 試料をJIS K 0121の8.(操作方法)の操作に準じてフレーム中に噴霧し,波長589.0 nmの指示値(吸
光度又はその比例値)を読み取る。
b) 空試験として,この操作に用いた水について,a) の操作を行って試料について得た指示値を補正する。
c) あらかじめ,次によって作成した検量線からナトリウムの濃度を求め,試料中のナトリウムの濃度
(mg/L)を算出する。
1) ナトリウム標準液(Na:10 mg/L)0.5〜40 mLを全量フラスコ100 mLに段階的にとり,水を標線ま
で加える。これらの溶液についてa) の操作を行う。
2) 別に,空試験としてこの操作に用いた水についてa) の操作を行ってそれぞれの標準液について得
た指示値を補正した後,ナトリウム(Na)の濃度と指示値との関係線を作成する。検量線の作成は
試料測定時に行う。
21.3.4
留意事項
a) 懸濁物質が含まれる場合は,21.2.4 a) の操作を行う。
b) 試料がアルカリ性の場合は,21.2.4 b) の操作を行う。
c) カリウム,カルシウムが共存すると正の誤差を生じる。このような試料には,21.2.4 d) の操作を行う。
21.4
電気加熱原子吸光法
試料を電気加熱炉で原子化し,ナトリウムによる原子吸光を波長589.0 nmで測定してナトリウムを定量
する。
定量範囲:Na:1〜20 µg/L,20〜100 µg/L,繰返し精度:5〜20 %(装置及び測定条件によって異なる。)
この方法は,共存する酸,塩の種類及び濃度の影響を受けやすいので,これらの影響の少ない試料(例
えば,貫流ボイラの給水,揮発性物質処理を行っている水管ボイラの給水及びボイラ水の試料)に適用す
る。
21.4.1
試薬
試薬は,次による。
a) 水 JIS K 0557で規定するA4の水。定量する元素について空試験を行って使用に支障がないことを
96
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
確認しておく。
b) ナトリウム標準液(Na:1 mg/L) 21.3.1 c) のナトリウム標準液(Na:10 mg/L)20 mLを全量フラ
スコ200 mLにとり,水を標線まで加える。使用時に調製する。
21.4.2
器具及び装置
器具及び装置は,次による。
a) 電気加熱原子吸光分析装置 JIS K 0121で規定する電気加熱原子吸光分析装置で,測定対象元素用の
光源を備え,かつ,バックグラウンド補正が可能なもの。
b) マイクロピペット JIS K 0970で規定するピストン式ピペット10〜50 μL,又は自動注入装置。
21.4.3
操作
操作は,次による。
a) 試料の一定量(10〜50 μL)をマイクロピペットで発熱体に注入し,JIS K 0121の8.(操作方法)の操
作に準じて,乾燥(100〜120 ℃,30〜40秒間)した後,灰化(600〜1 000 ℃,30〜40秒間)し,次
に,原子化(2 200〜2 700 ℃,3〜6秒間)し,波長589.0 nmの指示値を読み取る。この操作を3回
繰り返し,指示値が合うことを確認する。
なお,乾燥,灰化,原子化の温度及び時間は用いる装置,試料の注入量及び共存する塩類の濃度に
よって異なるので,装置や試料によってこれらの条件の最適化をはかる。
b) 空試験として,a) の操作を行って試料について得た指示値を補正する。
c) あらかじめ,次によって作成した検量線からナトリウムの濃度を求め,試料中のナトリウムの濃度
(μg/L)を算出する。
1) ナトリウム標準液(Na:1 mg/L)0.1〜2 mL[又はナトリウム標準液(Na:1 mg/L)2〜10 mL]を
全量フラスコ100 mLに段階的にとり,水を標線まで加える。これらの溶液についてa) の操作を行
う。
2) 別に,空試験としてb) の操作を行ってそれぞれの標準液について得た指示値を補正する。
3) ナトリウム(Na)の濃度と指示値との関係線を作成する。検量線の作成は,試料測定時に行う。
21.4.4
留意事項
a) 懸濁物質が含まれる場合は,21.2.4 a) の操作を行う。
b) 試料がアルカリ性の場合は,21.2.4 b) の操作を行う。
c) 試料中のナトリウム濃度が極低濃度の場合には,試料の一定量(10〜50 μL)を発熱体に注入し,乾燥
する。この操作を繰り返した後,灰化,原子化の操作を行って指示値を読み取る。この場合は,21.4.3
b) の操作でも同様な回数を注入し,乾燥する。
21.5
ICP発光分光分析法
試料を試料導入部を通して誘導結合プラズマ中に噴霧し,ナトリウムによる発光強度を波長589.592 nm
で測定して,ナトリウムを定量する。この方法によって,ナトリウムのほかに表9に示す元素が同時定量
できる。それぞれの元素の測定波長,定量範囲,繰返し精度の例を表9に示す。
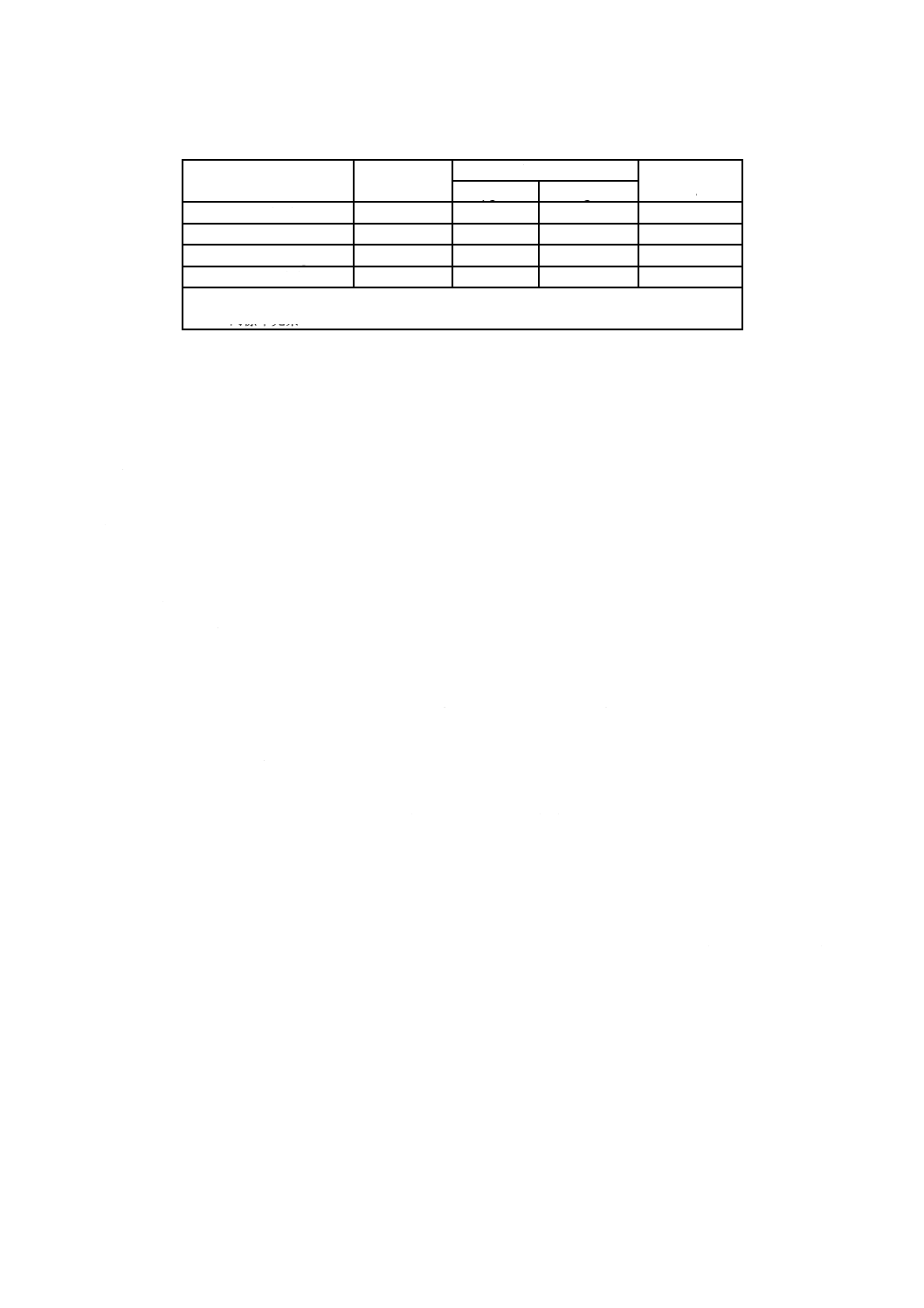
97
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表9−対象元素の定量範囲などの例a)
対象元素
測定波長
nm
定量範囲
繰返し精度
%
μg/L
mg/L
ナトリウム(Na)
589.592
−
0.5〜10
2〜10
カルシウム(Ca)
393.367
10〜200
0.2〜5
2〜10
マグネシウム(Mg)
279.553
5〜100
0.1〜3
2〜10
イットリウム(Y)b)
371.029
−
−
−
注a) 装置及び測光方式,測定条件によって異なる。
b) 内標準元素
ナトリウムの測定は,共存する酸,塩の種類及び濃度の影響を受けやすいので,これらの影響の少ない
試料(例えば,貫流ボイラの給水,揮発性物質処理を行っている水管ボイラの給水及びボイラ水の試料)
に適用する。
21.5.1
試薬
試薬は,次による。
a) 水 21.2.1 a) による。
b) 塩酸(1+1) JIS K 8180で規定する塩酸を用いて調製する。
c) ナトリウム標準液(Na:1 000 mg/L) 21.2.1 b) による。
d) カルシウム標準液(Ca:1 000 mg/L) 国家計量標準にトレーサブルなカルシウム標準液(Ca:1 000
mg/L)を使用するか,又は,次による。JIS K 8617で規定する炭酸カルシウムを105±2 ℃で約2時
間加熱し,デシケータ中で放冷する。その2.498 gをとり,水約50 mLに分散させ,これに塩酸(1+
1)20 mLを加えて溶かす。沸騰しない程度に数分間加熱して二酸化炭素を除く。放冷後,全量フラス
コ1 000 mLに移し入れ,水を標線まで加える。
e) マグネシウム標準液(Mg:1 000 mg/L) 国家計量標準にトレーサブルなマグネシウム標準液(Mg:
1 000 mg/L)を使用するか,又は,次による。JIS K 8432で規定する酸化マグネシウムを約800 ℃で
約2時間加熱し,デシケータ中で放冷する。その1.658 gを塩酸(1+1)20 mLに溶かして全量フラス
コ1 000 mLに移し入れ,水を標線まで加える。
f)
イットリウム溶液(Y:1 000 mg/L) 酸化イットリウム(III)0.318 gをとり,JIS K 9901で規定す
る高純度試薬−硝酸5 mLを加え,加熱して溶かし,煮沸して窒素酸化物を追い出す。放冷後,全量
フラスコ250 mLに移し,水を標線まで加える。
g) イットリウム溶液(Y:50 mg/L) 使用時にイットリウム溶液(Y:1 000 mg/L)25 mLを全量フラ
スコ500 mLにとり,塩酸(1+1)10 mLを加え,水を標線まで加える。
h) 混合標準液[(Na:20 mg,Ca:20 mg,Mg:10 mg)/L] c) のナトリウム標準液(Na:1 000 mg/L)
10 mL,d) のカルシウム標準液(Ca:1 000 mg/L)10 mL,e) のマグネシウム標準液(Mg:1 000 mg/L)
5 mLを全量フラスコ500 mLにとり,塩酸(1+1)10 mLを加え,水を標線まで加える。使用時に調
製する。
21.5.2
器具及び装置
器具及び装置は,次による。
a) ICP発光分光分析装置 JIS K 0116による。
21.5.3
準備操作
準備操作は,次による。
98
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) 試料の適量を全量フラスコ100 mLにとり,塩酸の濃度が約0.1 mol/Lになるように塩酸(1+1)を加
え,水を標線まで加える。このときの濃度が表9の定量範囲になるようにする。
21.5.4
操作
操作は,次による。
a) 21.5.3 a) の溶液をJIS K 0116の4.7(定量分析)に準じて,試料導入部を通してプラズマ中に噴霧し,
波長589.592 nmの発光強度を測定する。
b) 空試験として塩酸(1+1)を水で薄めて塩酸の濃度を約0.1 mol/Lとした溶液についてa) の操作を行
って,試料について得た発光強度を補正する。
c) 検量線からナトリウムの濃度を求め,試料中のナトリウムの濃度(mg/L又はμg/L)を算出する。
d) ナトリウムと同時にカルシウム,マグネシウムを同時定量する場合には,a) 及びb) においてそれぞ
れの元素における波長の発光強度を測定し,c) と同様に次のように作成した検量線を用いて,それぞ
れの元素の濃度を算出する。
1) 混合標準液[(Na:20 mg,Ca:20 mg,Mg:10 mg)/L]0.05〜1 mL(又は1〜50 mL)を全量フラス
コ100 mLに段階的にとり,試料と同じ酸の濃度となるように塩酸(1+1)を加えた後,水を標線
まで加える。これらの溶液についてa) の操作を行う。
2) 別に,空試験としてこの操作に用いた水について試料と同じ条件になるように塩酸(1+1)を加え
た後,b) の操作を行ってそれぞれの標準液について得た発光強度を補正し,測定対象元素の濃度と
発光強度との関係線を作成する。検量線の作成は試料測定時に行う。
21.5.5
留意事項
a) 懸濁物質が含まれる場合は,21.2.4 a) の操作を行う。
b) 試料がアルカリ性の場合は,21.2.4 b) の操作を行う。
c) ナトリウムの定量において,多量のカリウム及びリチウムが共存する試料の場合は,21.2.4 d) による。
d) 波長の異なる2本以上のスペクトル線を同時測定が可能な装置では,内標準法は,次による。
1) 試料の適量を全量フラスコ100 mLにとり,21.5.1 g) のイットリウム溶液(Y:50 mg/L)10 mLを
加えた後,塩酸の濃度が約0.1 mol/Lになるように塩酸(1+1)を加え,水を標線まで加える。
2) 21.5.4 a) の操作を行って測定対象元素の波長と同時に測定対象元素の発光強度とイットリウムの
波長の発光強度を測定し,測定対象元素とイットリウムとの発光強度の比を求める。
3) 空試験として試料と同量の水を用いて,1) 及び2) の操作を行う。
4) 各測定対象元素の検量線から,試料中の測定対象元素の濃度を算出する。
4.1) 混合標準液[(Na:20 mg,Ca:20 mg,Mg:10 mg)/L]0.05〜1 mL(又は1〜50 mL)を全量フラ
スコ100 mLに段階的にとり,21.5.1 g) のイットリウム溶液(Y:50 mg/L)10 mLをそれぞれ加え,
塩酸の濃度が約0.1 mol/Lになるように塩酸(1+1)を加え,水を標線まで加える。これらの溶液
について2) の操作を行って測定対象元素の各濃度における測定対象元素とイットリウムとの発
光強度比を求める。
4.2) 別に,空試験として混合標準液に代えて水を用い,測定を行って測定対象元素の各濃度における
測定対象元素とイットリウムとの発光強度比を求め,測定対象元素の発光強度比を補正する。各
測定対象元素の濃度の発光強度比の関係線を作成し,検量線とする。
e) 高次のスペクトル線が使用可能な装置では,高次のスペクトル線を用いて測定してもよい。
f)
精度,正確さを確認してあれば,表9に示す以外の波長を用いてもよい。

99
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
21.6
ICP質量分析法
試料を前処理した後,内標準元素を加え,試料導入部を通して高周波プラズマ中に噴霧し,ナトリウム
及び内標準元素(イットリウム,インジウム又はビスマス)のそれぞれの質量/電荷数(m/z)における指
示値(イオンカウント値又はその比例値)を測定し,ナトリウムの指示値と内標準元素の指示値との比を
求めてナトリウムを定量する。この方法によって,ナトリウムのほかに表10に示す元素が同時定量でき
る。それぞれの元素ごとの定量範囲,繰返し精度などの例を,表10に示す。
表10−定量範囲,繰返し精度及び質量数の例a)
対象元素
定量範囲
繰返し精度
質量数
μg/L
%
ナトリウム(Na)
0.5〜500
5〜20
23
マグネシウム(Mg)
0.5〜500
5〜20
24,25,26
イットリウム(Y)b)
−
−
89
インジウム(In)b)
−
−
115
ビスマス(Bi)b)
−
−
209
注a) 装置及び測定条件によって異なる。
b) 内標準元素はいずれかを使用する。
21.6.1
試薬
試薬は,次による。
a) 水 21.4.1 a) による。
b) 硝酸(1+1) JIS K 9901で規定する高純度試薬−硝酸を用いて調製する。
c) ナトリウム標準液(Na:100 mg/L) 21.3.1 b) による。
d) マグネシウム標準液(Mg:100 mg/L) 21.5.1 e) のマグネシウム標準液(Mg:1 000 mg/L)10 mL
を全量フラスコ100 mLにとり,水を標線まで加える。
e) イットリウム溶液(Y:1 mg/L) 使用時に,21.5.1 f) のイットリウム溶液(Y:1 000 mg/L)1 mL
を全量フラスコ1 000 mLにとり,硝酸(1+1)20 mLを加え,水を標線まで加える。
f)
インジウム溶液(In:1 mg/L) 国家計量標準にトレーサブルなインジウム標準液(In:1 000 mg/L)
を準備し,使用時に,この溶液1 mLを全量フラスコ1 000 mLにとり,硝酸(1+1)20 mLを加え,
水を標線まで加えるか,又は,次による。インジウム0.250 gをとり,JIS K 9901で規定する高純度試
薬−硝酸10 mLを加え,加熱して溶かし,煮沸して窒素酸化物を追い出し,放冷後,全量フラスコ250
mLに移し入れ,水を標線まで加えた溶液を準備調製する。使用時に,この溶液1 mLを全量フラスコ
1 000 mLにとり,硝酸(1+1)20 mLを加え,水を標線まで加える。
g) ビスマス溶液(Bi:1 mg/L) 国家計量標準にトレーサブルなビスマス標準液(Bi:1 000 mg/L)を
準備し,この溶液1 mLを全量フラスコ1 000 mLにとり,硝酸(1+1)20 mLを加え,水を標線まで
加えるか,又は,次による。酸化ビスマス(III)0.279 gをとり,硝酸(1+1)10 mLを加え,加熱し
て溶かし,放冷後,全量フラスコ250 mLに移し入れ,水を標線まで加えた溶液を準備調製する。こ
の溶液1 mLを全量フラスコ1 000 mLにとり,硝酸(1+1)20 mLを加え,水を標線まで加える。
h) 混合標準液[(Na:1 mg,Mg:1 mg)/L] c) のナトリウム標準液(Na:100 mg/L)10 mL,d) の
マグネシウム標準液(Mg:100 mg/L)10 mLをそれぞれ全量フラスコ1 000 mLにとり,硝酸(1+1)
1.5 mLを加え,水を標線まで加える。使用時に調製する。ただし,金属元素のうち,定量する金属元
100
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
素の混合標準液又は単独の標準液を調製して使用してもよい。
21.6.2
器具及び装置
器具及び装置は,次による。
a) ICP質量分析計 JIS K 0133による。分子イオンのピークによる干渉を低減する方法としてプラズマ
の温度を低くする方式の装置がある。また,コリジョン・リアクションセルなどのスペクトル干渉除
去機能が搭載されている装置もある。この装置では,22.5に示す分析成分と同時測定を行うことがで
きる。
21.6.3
準備操作
準備操作は,次による。
a) 試料の適量を全量フラスコ100 mLにとり,いずれかの内標準溶液(1 mg/L)1 mLを加え,硝酸の最
終濃度が0.1〜0.5 mol/Lになるように硝酸(1+1)を加えた後,水を標線まで加える。このときの濃
度が表10の定量範囲になるようにする。
b) 空試験の溶液として同量の水を全量フラスコ100 mLにとり,a) と同じ内標準溶液(1 mg/L)1 mLを
加え,試料と同じ酸の濃度になるように硝酸(1+1)を加えた後,水を標線まで加える。
c) JIS K 0133の箇条8(分析装置の最適化),箇条9(分析条件の決定)に準じて,ICP質量分析計を定
量できる状態に調整する。
21.6.4
操作
操作は,次による。
a) 21.6.3 a) の試料をJIS K 0133の箇条12(定量分析)に準じて試料導入部を通して高周波プラズマ中
に噴霧して測定対象元素と内標準物質との質量/電荷数における指示値[目的元素の指示値(イオン
電流又はその比例値)]を読み取り,測定対象元素の指示値と内標準物質の指示値との比を求める。
b) 空試験として21.6.3 b) の溶液についてa) の操作を行って測定対象元素の指示値と内標準物質の指示
値との比を求め,試料について得た測定対象元素と内標準物質との指示値の比を補正する。
c) 検量線から測定対象元素の濃度を求め,試料中の測定対象元素の濃度(ng/L又はµg/L)を算出する。
d) ナトリウムとマグネシウムとを同時定量する場合には,各金属元素について,それぞれの検量線を作
成する。検量線の作成は,試料測定時に行う。
1) 21.6.3 a) の試料の代わりに混合標準液[(Na:1 mg,Mg:1 mg)/L]0.05〜50 mLを用い,検量線
用混合標準液を調製して検量線を作成する。これらの溶液についてa) の操作を行う。
2) 別に,空試験としてこの操作に用いた水を全量フラスコ100 mLにとり,内標準溶液(1 mg/L)1 mL
を加え,21.6.3 a) の試料と同じ酸の濃度になるように硝酸(1+1)を加えた後,同じ水を標線まで
加え,a) の操作を行ってそれぞれの標準液について得た指示値の比を補正し,測定対象元素の指示
値と内標準元素の指示値との比の関係線を作成する。
21.6.5
留意事項
a) 懸濁物質が含まれる場合は,21.2.4 a) の操作を行う。
b) 試料がアルカリ性の場合は,21.2.4 b) の操作を行う。
c) 各元素の質量数を設定するには,表10を参考するとよい。
d) 妨害物質の存在が不明の場合には,定量に先立って高周波プラズマ質量分析装置による定性分析を行
うことによって目的とする元素及び内標準物質の測定質量数に対する妨害を推定することができる。
妨害が認められる場合には,内標準物質の変更,試料の希釈又は前処理を行って妨害の低減を図る。
妨害物質の影響が無視できる試料の場合は,内標準物質の添加を省略して検量線法によって定量して
101
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
もよい。
e) 安定同位体がある場合,複数の同位体の質量/電荷数を用いて測定を行うことによってスペクトル干
渉による妨害を推定することができる。スペクトル干渉による影響が無視できない場合は,試料をさ
らに薄めて測定する。それでも影響を受ける場合は,適切な分離方法を用いて妨害となるマトリック
スを除去した後,測定を行う。
f)
定量下限付近の定量で,水による指示値が無視できない場合には,使用する水を更に精製して試験に
支障のないものにする。
21.7
イオン電極法
試料のpHを10.2〜10.6に調節し,ナトリウムイオン電極を指示電極として電位を測定し,ナトリウム
を定量する。
定量範囲:Na:1〜100 mg/L,繰返し精度:5〜20 %(装置及び測定条件によって異なる。)
21.7.1
試薬
試薬は,次による。
a) 水 21.2.1 a) による。
b) トリス緩衝液 JIS K 9704で規定する2-アミノ-2-ヒドロキシメチル-1,3-プロパンジオール60 gをと
り,水に溶かして1 Lとする。
c) 酢酸リチウム溶液(1 mol/L) 酢酸リチウム二水和物を用いて調製する。
d) 酢酸リチウム緩衝液 酢酸リチウム二水和物102 gを水約500 mLに溶かし,これにJIS K 8355で規
定する酢酸2 mLと水を加えて全量を1 Lとする。
e) ナトリウム標準液(Na:1 000 mg/L) 21.2.1 b) による。
f)
ナトリウム標準液(Na:100 mg/L) 21.3.1 b) による。
g) ナトリウム標準液(Na:10 mg/L) 21.3.1 c) による。
h) ナトリウム標準液(Na:5 mg/L) ナトリウム標準液(Na:10 mg/L)10 mLを全量フラスコ200 mL
にとり,水を標線まで加える。使用時に調製する。
i)
ナトリウム標準液(Na:1 mg/L) ナトリウム標準液(Na:10 mg/L)20 mLを全量フラスコ200 mL
にとり,水を標線まで加える。使用時に調製する。
j)
アンモニア水(1+1) JIS K 8085で規定するアンモニア水を用いて調製する。
k) 塩酸(1+2) JIS K 8180で規定する塩酸を用いて調製する。
21.7.2
器具及び装置
器具及び装置は,次による。
a) 電位差計 15.5.2 a) による。
b) 指示電極 JIS K 0122で規定するガラス膜又は液体膜のナトリウムイオン電極。標準液を用いた起電
力の応答は,25 ℃におけるナトリウム濃度の10倍濃度変化当たり,55 mV以上,液温10〜30 ℃で
の応答時間は,2〜3分間以内のもの。また,ナトリウム標準液(Na:1〜100 mg/L)の範囲で直線の
検量線が得られるもの。ナトリウム標準液(Na:1 mg/L)とナトリウム(Na:100 mg/L)との電位の
差は,100〜120 mV(25 ℃)の範囲に入る。
c) 参照電極 15.5.2 d) による。ただし,指示電極にガラス膜型電極を用いる場合には,この参照電極の
外筒液に硝酸アンモニウム溶液(100 g/L)又は硝酸カリウム溶液(100 g/L)を,液体膜型電極を用い
る場合には,酢酸リチウム溶液(1 mol/L)又は酢酸リチウム緩衝液を用いる。
d) イオン濃度計 15.5.2 b) による。
102
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
e) マグネチックスターラ 15.5.2 g) による。
21.7.3
準備操作
準備操作は,次による。
a) ナトリウムイオン電極をナトリウム標準液(Na:1 mg/L)に浸し,指針が安定していることを確認す
る。
b) ナトリウム標準液(Na:1 mg/L)100 mLをビーカ200 mLにとり,ガラス膜型電極の場合はトリス緩
衝液10 mLを,液体膜型電極の場合は酢酸リチウム緩衝液10 mLを加える。
c) これに指示電極と参照電極とを浸し,マグネチックスターラで泡が電極に触れない程度に強くかき混
ぜる。
d) 液温をはかり,電位差計で電位を測定する。
e) ガラス膜型電極の場合はナトリウム標準液(Na:10 mg/L)100 mL及びナトリウム標準液(Na:100 mg/L)
100 mLを,それぞれビーカ200 mLにとり,トリス緩衝液10 mLを加える。液体膜電極を用いる場合
は,ナトリウム標準液100 mLにつき酢酸リチウム緩衝液10 mLを加え,pHを10.2〜10.6に調節する。
それぞれの溶液の液温をd) での液温の±1 ℃に調節し,c) 及びd) の操作を行ってナトリウム標準
液(Na:10 mg/L,100 mg/L)の電位を測定する。
f)
片対数方眼紙の対数軸にナトリウムの濃度(mg/L)を,均等軸に電位(mV)をとって,ナトリウム
の濃度と電位との関係線を作成する。
21.7.4
操作
21.7.4.1 ガラス膜又は液体膜のナトリウムイオン電極を用いる場合
操作は,次による。
a) 試料100 mLをビーカ200 mLにとり,トリス緩衝液又は酢酸リチウム緩衝液10 mLを加え,液温を
21.7.3 d) での液温の±1 ℃に調節する。ただし,試料が酸性又は強アルカリ性で,トリス緩衝液を加
えてもpHが10.2〜10.6の範囲に入らない場合には,試料の適量を全量フラスコ200 mLにとり,これ
にアンモニア水(1+1)又は塩酸(1+2)を加えて,pHを約10に調節した後,水を標線まで加えた
ものから100 mLをとる。ナトリウム濃度が100 mg/L以上の場合は,試料の適量をとり,水で100 mL
にしたものを用いる。
b) 21.7.3のc) 及びd) の操作を行って電位を測定する。検量線からナトリウムの濃度を求め,試料中の
ナトリウムの濃度(mg/L)を算出する。
なお,a) で試料を希釈した場合には,希釈倍率に応じて濃度を補正する。
注記 ガラス膜型及び液体膜型のナトリウムイオン電極における主な共存物質の許容限度をナトリ
ウムイオンに対する化学当量最大比率で示す。
ガラス膜型 Li+:4.5×10,K+:4×102,NH4+:1.3×103,Ag+:0.2
液体膜型
K+:1.25×102,Li+:6.25×103,NH4+:1.4×103,Ca2+:1.5×104
界面活性剤が共存するとドリフトが生じるので注意する。
21.7.4.2 イオン濃度計を用いる場合
操作は,次による。
a) ナトリウム標準液(Na:1 mg/L),ナトリウム標準液(Na:100 mg/L)100 mLを別々のビーカにとり,
それぞれトリス緩衝液又は酢酸リチウム緩衝液を10 mL加える。
b) 液温を測定する。
c) 21.7.3 c) の操作を行って,イオン濃度計の指示値をNa:1 mg/LとNa:100 mg/Lになるように調節す
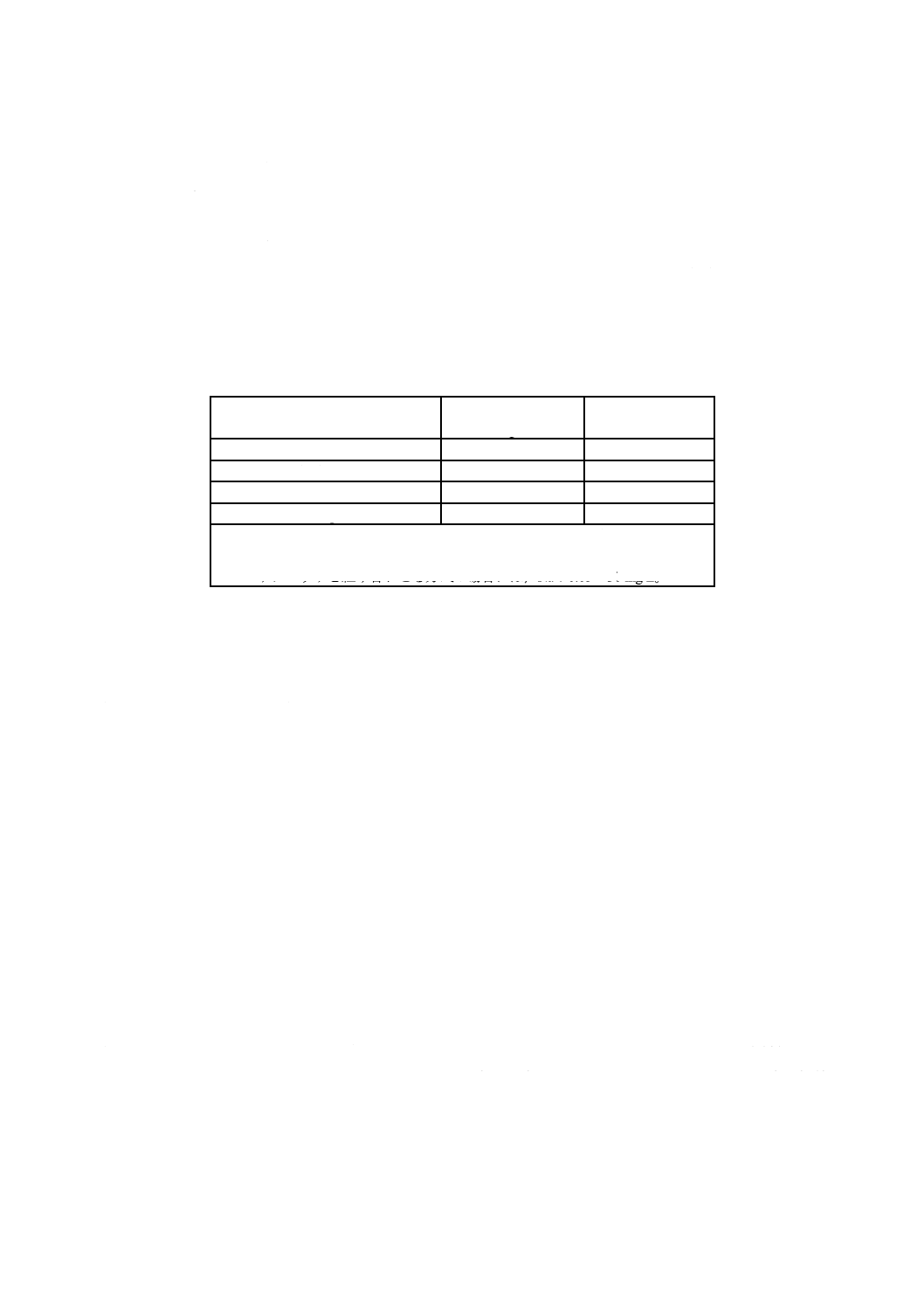
103
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
る。
d) ナトリウム標準液(Na:5 mg/L)を用いて,イオン濃度計の指示値を確認する。
e) 21.7.4.1 a) で処理した試料をビーカにとり,イオン濃度計の指示値を読み,試料のナトリウム濃度
(mg/L)を求める。
21.8
イオンクロマトグラフ法
試料中の陽イオンをイオンクロマトグラフ法によって定量する。検出器には,電気伝導率検出器を用い
る。この方法によって,ナトリウムのほかに表11に示す陽イオンが同時定量できる。それぞれの陽イオン
の定量範囲及び繰返し精度の例を,表11に示す。
表11−各陽イオンの定量範囲及び繰返し精度の例a)
対象陽イオン
定量範囲
mg/L
繰返し精度
%
アンモニウムイオン(NH4+)
0.1〜30
2〜10
ナトリウム(Na)b)
0.1〜30
2〜10
カルシウム(Ca)
0.2〜50
5〜10
マグネシウム(Mg)
0.2〜50
5〜10
注a) 定量範囲は,検出器,試料注入量,カラムのイオン交換容量などによっ
て変わる。
b) サプレッサと組み合わせる方式の場合には,Na:0.05〜30 mg/L。
21.8.1
試薬
試薬は,次による。
a) 水 21.2.1 a) による。
b) 塩酸(1+1) 21.5.1 b) による。
c) 溶離液 溶離液は,装置の種類及び分離カラムに充塡した陽イオン交換体の種類によって異なるので,
あらかじめ,アンモニウムイオン,ナトリウム,カルシウム及びマグネシウムの各イオンの分離の確
認を,21.8.5 a) によって行う。
d) 再生液 再生液は,サプレッサを用いる場合に使用するが,装置の種類及びサプレッサの種類によっ
て再生液が異なる。あらかじめ分離カラムと組み合わせて,21.8.5 a) の操作を行って再生液の性能を
確認する。
e) アンモニウムイオン標準液(NH4+:1 000 mg/L) 国家計量標準にトレーサブルなアンモニウムイオ
ン標準液(NH4+:1 000 mg/L)を使用するか,又は,次による。JIS K 8116で規定する塩化アンモニ
ウムをデシケータ中[JIS K 8228で規定する過塩素酸マグネシウム(乾燥用)を入れたもの。]に16
時間以上放置し,その2.97 gをとり,少量の水に溶かし全量フラスコ1 000 mLに移し入れ,水を標線
まで加える。
f)
ナトリウム標準液(Na:1 000 mg/L) 21.2.1 b) による。
g) カリウム標準液(K:1 000 mg/L) 国家計量標準にトレーサブルなカリウム標準液(K:1 000 mg/L)
を使用するか,又は,次による。JIS K 8121で規定する塩化カリウムを500〜600 ℃で約1時間加熱
し,デシケータ中で放冷する。その0.191 gをとり,少量の水に溶かし,全量フラスコ100 mLに移し
入れ,水を標線まで加える。
h) カルシウム標準液(Ca:1 000 mg/L) 21.5.1 d) による。
104
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
i)
マグネシウム標準液(Mg:1 000 mg/L) 21.5.1 e) による。
j)
陽イオン混合標準液[(NH4+:100 mg,Na:100 mg,K:100 mg,Ca:200 mg,Mg:200 mg)/L] ア
ンモニウムイオン標準液(NH4+:1 000 mg/L)10 mL,ナトリウム標準液(Na:1 000 mg/L)10 mL,
カリウム標準液(K:1 000 mg/L)10 mL,カルシウム標準液(Ca:1 000 mg/L)20 mL及びマグネシ
ウム標準液(Mg:1 000 mg/L)20 mLをそれぞれ全量フラスコ100 mLにとり,水を標線まで加える。
使用時に調製する。
k) 陽イオン混合標準液[(NH4+:10 mg,Na:10 mg,K:10 mg,Ca:20 mg,Mg:20 mg)/L] アン
モニウムイオン標準液(NH4+:1 000 mg/L)5 mL,ナトリウム標準液(Na:1 000 mg/L)5 mL,カリ
ウム標準液(K:1 000 mg/L)5 mL,カルシウム標準液(Ca:1 000 mg/L)10 mL及びマグネシウム標
準液(Mg:1 000 mg/L)10 mLをそれぞれ全量フラスコ500 mLにとり,水を標線まで加える。また
は,陽イオン混合標準液[(NH4+:100 mg,Na:100 mg,K:100 mg,Ca:200 mg,Mg:200 mg)/L]
10 mLを全量フラスコ100 mLにとり,水を標線まで加える。いずれも使用時に調製する。
21.8.2
器具及び装置
器具及び装置は,次による。
a) イオンクロマトグラフ イオンクロマトグラフには,分離カラムとサプレッサとを組み合わせた方式
のものと,分離カラム単独の方式のものとがある。いずれでもよいが,次に掲げる条件を満たすもの
で,アンモニウムイオン,ナトリウム,カリウム,カルシウム及びマグネシウムの各イオンが分離定
量できるもの。
1) 分離カラム ステンレス鋼製又は四ふっ化エチレン樹脂製,ポリエーテルエーテルケトン製などの
合成樹脂製のものに,陽イオン交換体を充塡したもの。
2) サプレッサ 溶離液中の陰イオンの濃度に対して十分なイオン交換能力をもつ陰イオン交換膜(膜
形,電気透析形がある。)又は同様な性能をもった陰イオン交換体を充塡したもの。再生液と組み合
わせて用いる。
3) 検出器 電気伝導率検出器
4) データ処理部 JIS K 0127の5.7(データ処理部)による。
21.8.3
準備操作
準備操作は,次による。
a) 試料を15.6.3 a) に従ってろ過する。必要に応じて15.6.3 b) に従って希釈する。
b) 分離カラムの性能の確認は定期的に21.8.5 a) の操作によって行う。
21.8.4
操作
操作は,次による。
a) イオンクロマトグラフ装置を定量できる状態にし,分離カラムに溶離液を一定の流量(例えば,1〜2
mL/min)で流しておく。サプレッサを必要とする装置では再生液を一定の流量で流しておく。
b) 21.8.3 a) の準備操作を行った試料の一定量(例えば,50〜200 μLの一定量)をイオンクロマトグラフ
装置に注入してクロマトグラムを記録する。
c) クロマトグラム上のナトリウムに相当するピークについて,ピーク高さ又はピーク面積(指示値)を
読み取る。
d) 試料を薄めた場合には,空試験として試料と同量の水について,b) 及びc) の操作を行って試料につ
いて得た指示値を補正する。
e) あらかじめ,次によって作成した検量線からナトリウムの濃度を求め,試料中のナトリウムの濃度
105
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(mg/L)を算出する。
1) ナトリウム標準液(Na:100 mg/L)0.1〜30 mLを段階的に全量フラスコ100 mLにとり,水を標線
まで加え,b) 及びc) の操作を行ってそれぞれのナトリウムに相当する指示値(ピーク高さ又はピ
ーク面積)を読み取る。
2) 別に,空試験として水についてb) 及びc) の操作を行ってそれぞれのナトリウムに相当する指示値
を補正した後,ナトリウム(Na)の濃度と指示値との関係線を作成する。検量線の作成は測定時に
行う。
3) ナトリウムと他の陽イオン成分を同時に測定する場合は,陽イオン混合標準液を用いて測定成分ご
とに検量線を作成する。
21.8.5
留意事項
a) 溶離液を一定の流量(例えば,1〜2 mL/min)で流し,陽イオン混合標準液[(NH4+:10 mg,Na:10
mg,K:10 mg,Ca:20 mg,Mg:20 mg)/L]の一定量をイオンクロマトグラフに注入し,クロマトグ
ラムを求め,各陽イオンが分離できるものを用いる。イオンクロマトグラフの性能として分離度(R)
が1.3以上のものを用いる。分離度を求めるには,溶離液を一定の流量(例えば,1〜2 mL/min)で流
す。クロマトグラムのピーク高さがほぼ同程度となるような濃度の陽イオン混合溶液を調製して,ク
ロマトグラムを作成し,次の式によって算出する。
(
)
2
1
1
2
2
W
W
t
t
R
R
R
+
−
×
=
ここに,
tR1: 第1ピークの保持時間(s)
tR2: 第2ピークの保持時間(s)
W1: 第1ピークのピーク幅(s)
W2: 第2ピークのピーク幅(s)
b) 分離カラムは,使用を続けると性能が低下するので,定期的にa) の操作で確認する。性能が低下し
ている場合には,溶離液の20〜200倍の濃度の溶液を調製し,分離カラムに注入し,洗浄した後,性
能を確認する。性能が回復しない場合は新品と取り替える。
c) 分離カラムの性能は,試料中の懸濁物,有機物などによっても汚染されて性能が徐々に低下する。懸
濁物を含む試料は,21.8.3 a) の準備操作で除去した後,試験する。また,有機物(たん白質,油脂,
界面活性剤など)を含む試料は限外ろ過膜でろ過し,できるだけ有機物を除去した後,試験する。
d) 酸化性物質又は還元性物質が共存すると,分離カラムの分離性能が低下する。このような場合には,
試料を水で一定の割合に薄めて試験すれば,ある程度は影響を防ぐことができる。
e) カルボン酸形の陽イオン交換カラムと,溶離液として硝酸溶液,メタンスルホン酸溶液,[2,6-ピリジ
ンジカルボン酸-L(+)-酒石酸]溶液などを用いると,1価陽イオンのほかにカルシウム,マグネシウ
ムなど2価の陽イオンの溶離及び同時定量が可能になる。
f)
ナトリウムの濃度が1 mg/Lのときアンモニウムイオン及びカリウムは,いずれも100 mg/L以下なら
ば妨害しない。
21.9
ナトリウム(Na)プロセス用分析装置(イオン電極法)による測定方法
プロセス用分析装置(イオン電極法)によって給水のナトリウム濃度を連続的に測定する。
定量範囲:Na:0.1〜10 μg/L,1〜100 μg/L,繰返し精度:最大目盛値の±20 %
21.9.1
試薬
試薬は,次による。

106
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) 水 21.4.1 a) による。
b) pH調整液 それぞれの計測器の取扱説明書によって調製する。pH調整液には,アンモニア水(1+1),
2-アミノエタノール溶液(500 g/L),2-アミノ-2-ヒドロキシメチル-1,3-プロパンジオール溶液(60 g/L)
などを用いる。測定時のpHを10.2〜10.6に調整する。
c) スパン校正液 ナトリウム標準液(Na:10 mg/L)を用いて,測定レンジの25,50,75 %に相当する
校正液を調製する。
21.9.2
器具及び装置
器具及び装置は,次による。
a) 構成 構成を,図28に示す。
図28−プロセス用分析装置の構成
21.9.3
測定
測定は,次による。
a) 測定準備 あらかじめ,電極を12時間以上水に浸した後,そのままの状態で指示部に接続する。計測
器の各部を点検し,特に水漏れがないことを確認してから所定の手順に従って電源を入れ,装置が安
定するまで30分間以上待つ。
b) ゼロ校正 計測器が安定状態に達した後,電極を水(ゼロ校正液)に浸しゼロ調整を行う。又は,電
気的にゼロ調整を行う。ゼロ校正は,測定開始時及びpH調整液を調製し直したときなどに行う。
c) スパン校正 電極を測定レンジの25,50,75 %に相当するスパン校正液に浸し,指示値が安定した後,
スパン校正を行う。測定開始時,及び連続測定の場合は2〜4週間に1回程度の割合で行う。
d) 測定 電極を試料で十分に洗浄した後,試料溶液を設定流量で流す。指示値が安定した後に連続測定
を行う。
22
カルシウム(Ca)
22.1
一般事項
カルシウムの定量には,キレート滴定法,フレーム原子吸光法,ICP発光分光分析法,ICP質量分析法
又はイオンクロマトグラフ法を適用する。
22.2
キレート滴定法
試料のpHを12以上とし,指示薬としてHSNN[2-ヒドロキシ-1-[(2'-ヒドロキシ-4'-スルホ-1'-ナフタレ
ニル)アゾ]-3-ナフタレンカルボン酸(IUPACによる名称)]を加え,エチレンジアミン四酢酸二水素二
ナトリウム溶液で滴定してカルシウムを定量する。
定量範囲:Ca:1 mg/L以上,繰返し精度:2〜10 %
22.2.1
試薬
107
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
試薬は,次による。
a) 水 21.2.1 a) による。
b) 水酸化カリウム溶液 JIS K 8574で規定する水酸化カリウム250 gを水に溶かして500 mLとする。ポ
リエチレン瓶に保存する。
c) シアン化カリウム溶液(100 g/L) 10.2.1.1 a) による。
警告 シアン化カリウムは有毒である。取扱い及び廃液の処理は,法令に従い,安全及び健康に対
する適切な措置を取らなければならない。シアン化カリウムを含む溶液は,有毒なシアン化
水素ガスが発生するため,酸性にしてはならない。
d) 塩化ヒドロキシルアンモニウム溶液(100 g/L) 10.2.1.1 b) による。
e) HSNN溶液 JIS K 8776で規定する2-ヒドロキシ-1-(2-ヒドロキシ-4-スルホ-1-ナフチルアゾ)-3-ナ
フトエ酸0.5 gをJIS K 8891で規定するメタノール100 mLに溶かし,JIS K 8201で規定する塩化ヒド
ロキシルアンモニウム0.5 gを加える。着色ガラス瓶に保存する。
f)
HSNN粉末試薬 JIS K 8776で規定する2-ヒドロキシ-1-(2-ヒドロキシ-4-スルホ-1-ナフチルアゾ)-3-
ナフトエ酸0.5 gとJIS K 8962で規定する硫酸カリウム10 gとをめのう乳鉢で均一になるまでよくす
り潰し混合する。
g) 10 mmol/L EDTA溶液 10.2.1.1 e) による。この溶液1 mLは,Ca 0.400 8 mgに相当する。
22.2.2
操作
操作は,次による。
a) 懸濁物質が含まれている場合には,ろ過又は遠心分離によって除去した試料の適量(Caとして5 mg
以下を含む。)をビーカにとり,水を加えて約50 mLとする。
b) 水酸化カリウム溶液4 mLを加え,よくかき混ぜた後,約5分間放置する。放置した際に生じる沈殿
の量が多いと,終点が不明瞭になる。このような場合には1回の滴定で,おおよその滴定量を求めて
おき,別のビーカに同量の試料をとり,1回目の滴定に要した10 mmol/L EDTA溶液の量よりも1 mL
少ない量の10 mmol/L EDTA溶液を加え,水酸化カリウム溶液4 mLを加えてよく振り混ぜた後,約5
分間放置する。
c) シアン化カリウム溶液(100 g/L)0.5 mL及び塩化ヒドロキシルアンモニウム溶液(100 g/L)0.5 mL
を加えて,かき混ぜる。
d) 指示薬としてHSNN溶液5,6滴又はHSNN粉末約0.1 gを加え,すぐに10 mmol/L EDTA溶液で,溶
液の色が赤紫から青になるまで滴定する。
e) 次の式によって試料中のカルシウムの濃度(mg/L)を算出する。
8
400
.0
000
1
×
×
=
V
a
C
ここに,
C: カルシウム濃度(mg/L)
a: 滴定に要した10 mmol/L EDTA溶液(mL)
V: 試料(mL)
0.400 8: 10 mmol/L EDTA溶液1 mLのカルシウム相当量(mg)
22.2.3
留意事項
a) アルミニウム,バリウム,鉛,鉄,コバルト,銅,マンガン,すず及び亜鉛の金属イオンは,カルシ
ウムイオンと同じように滴定されるが,シアン化カリウム溶液の添加によって,亜鉛,銅及びコバル
トの各イオン妨害は最小になる。また,トリエタノールアミンを添加すると,アルミニウムイオンの
108
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
妨害は小さくなる。これらの金属イオンの共存は,終点の色の変化を不明瞭にするので,このような
場合は,22.2.2 b) の操作を行う。
b) オルトりん酸イオンの濃度が1 mg/L以上の場合は,滴定時のpH条件ではカルシウムイオンと反応し
て沈殿を生じる。
c) 滴定に時間がかかった場合,又はカルシウムの含有量が高い場合(100 mg/L又は2.5 mmol/Lより高い)
には,炭酸カルシウムの沈殿が生じる。
d) 鉄30 mg/L以下の妨害はシアン化カリウム250 mg又はJIS K 8663で規定する2,2',2''-ニトリロトリエ
タノール(トリエタノールアミン)5〜7 mLを滴定直前に添加することによって,マスキングできる。
22.2.1 e) HSNN溶液はメタノール溶液として用いるが,安定剤として塩化ヒドロキシルアンモニウム
を加えると,その還元作用及び溶液を酸性にする作用によって酸化が抑制される。
e) 水酸化カリウム溶液中に炭酸イオンが混在すると,試料中のカルシウムイオンが炭酸カルシウムとな
って沈殿する。この沈殿は,滴定の進行とともに溶けるが,滴定の反応を遅くする傾向がある。
f)
試料中にマグネシウムが多量に共存すると,多量の水酸化マグネシウムの沈殿が生成する。この場合
は,炭酸カルシウムを包み込むため,さらに滴定反応を遅らせ妨害する。また,指示薬HSNNの吸着
も多くなり,終点の判別が困難となることもある。このような影響をできるだけ少なくするため,第
1回の滴定を予備試験とし,22.2.2 b) のようにほぼ当量に近い量のEDTA溶液を加えることによって
炭酸カルシウムの生成を防ぐ。また,HSNN溶液の添加も滴定が終点付近に達した状態で行うことに
なり,その吸着を少なくし,終点の判別を容易にする。
g) 試料中にナトリウムが多量に共存するとEDTAは弱く結合するため,終点が判別しにくくなる。
22.3
フレーム原子吸光法
試料をアセチレン-空気フレーム中に噴霧し,カルシウムによる原子吸光を波長422.7 nmで測定してカ
ルシウムを定量する。
定量範囲:Ca:0.2〜4 mg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
22.3.1
試薬
試薬は,次による。
a) 水 21.2.1 a) による。
b) 塩酸(1+1) 21.5.1 b) による。
c) ランタン(III)溶液(50 g/L) 酸化ランタン(III)29 gを少量ずつ塩酸(1+1)500 mLに加えて溶
かす。
d) カルシウム標準液(Ca:1 000 mg/L) 21.5.1 d) による。
e) カルシウム標準液(Ca:10 mg/L) カルシウム標準液(Ca:1 000 mg/L)10 mLを全量フラスコ1 000
mLにとり,塩酸(1+1)10 mLを加えた後,水を標線まで加える。
22.3.2
器具及び装置
器具及び装置は,次による。
a) フレーム原子吸光分析装置 JIS K 0121で規定するフレーム原子吸光分析装置で,測定対象元素用の
光源を備え,かつ,バックグラウンド補正が可能なもの。
22.3.3
準備操作
準備操作は,次による。
a) 試料の適量を全量フラスコ100 mLにとり,塩酸(1+1)2 mLを加えた後,水を標線まで加える。こ
のときの濃度が0.2〜4 mg/Lの定量範囲になるようにする。
109
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) この溶液10 mLを乾いたビーカにとり,ランタン(III)溶液(50 g/L)1 mLを加える。
c) 空試験として試料と同量の水をとり,a) 及びb) の操作を行う。
22.3.4
操作
操作は,次による。
a) 22.3.3 b) の溶液をJIS K 0121の8.(操作方法)の操作に準じてフレーム中に噴霧し,波長422.7 nm
の指示値(吸光度又はその比例値)を読み取る。
b) 空試験として22.3.3 c) の溶液を用いてa) の操作を行って試料について得た指示値を補正する。
c) あらかじめ,次によって作成した検量線から試料中のカルシウムの濃度(mg/L)を算出する。
1) カルシウム標準液(Ca:10 mg/L)2〜40 mLを全量フラスコ100 mLに段階的にとり,試料と同じ
酸の濃度になるように塩酸(1+1)を加えた後,水を標線まで加える。これらの溶液について22.3.3
b) の操作を行った後,a) の操作を行う。
2) 別に,空試験としてこの操作に用いた水について試料と同じ酸の濃度になるように塩酸(1+1)を
加えた後,標準液と同様に操作して標準液について得た指示値を補正し,カルシウム(Ca)の濃度
と指示値との関係線を作成する。検量線の作成は試料測定時に行う。
22.3.5
留意事項
a) 懸濁物質が含まれる場合は,21.2.4 a) の操作を行う。
b) 試料がアルカリ性の場合は,21.2.4 b) の操作を行う。塩酸(1+1)の滴加量が多い場合には,測定し
たカルシウムの濃度(mg/L)を希釈割合に応じて補正する。
c) 多量のマグネシウム(1 000 mg/L以上)の共存は,負の誤差を与える。
d) りん酸塩,硫酸塩,アルミニウムなどは妨害するが,ランタン(III)溶液(50 g/L)を添加すること
によって抑制することができる。
22.4
ICP発光分光分析法
試料を試料導入部を通して誘導結合プラズマ中に噴霧し,カルシウムによる発光強度を波長393.367 nm
で測定して,カルシウムを定量する。この方法によって,カルシウムのほかに表9に示す元素が同時定量
できる。
22.4.1
試薬
試薬は,21.5.1による。
22.4.2
器具及び装置
器具及び装置は,21.5.2による。
22.4.3
準備操作
準備操作は,21.5.3による。
22.4.4
操作
操作は,21.5.4による。
22.4.5
留意事項
留意事項は,21.5.5による。
22.5
ICP質量分析法
試料を前処理した後,内標準元素(イットリウム,インジウム又はビスマス)を加え,試料導入部を通
して高周波プラズマ中に噴霧し,カルシウム及び内標準元素のそれぞれの質量/電荷数(m/z)における指
示値(イオンカウント値又はその比例値)を測定し,指示値と内標準元素の指示値との比を求めてカルシ
ウムを定量する。カルシウムの定量範囲,繰返し精度などの例を,表12に示す。
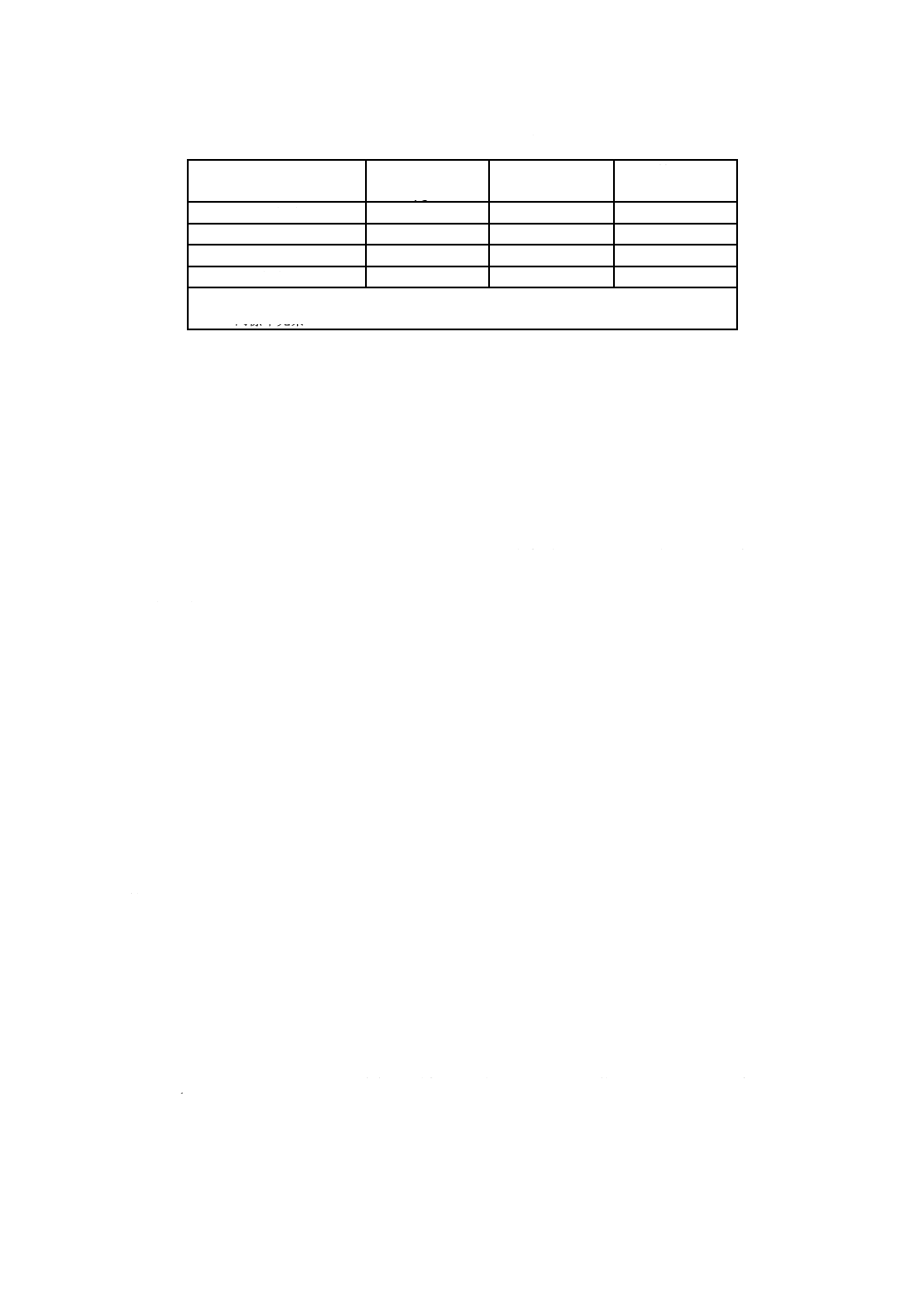
110
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表12−定量範囲,繰返し精度及び質量数の例a)
対象元素
定量範囲
繰返し精度
質量数
μg/L
%
カルシウム(Ca)
0.5〜500
2〜10
40,44
イットリウム(Y)b)
−
−
89
インジウム(In)b)
−
−
115
ビスマス(Bi)b)
−
−
209
注a) 装置及び測定条件によって異なる。
b) 内標準元素
22.5.1
試薬
試薬は,次による。
a) 水 21.4.1 a) による。
b) 硝酸(1+1) 21.6.1 b) による。
c) カルシウム標準液(Ca:1 000 mg/L) 21.5.1 d) による。
d) 内標準液(1 mg/L) 内標準元素としてイットリウム,インジウム又はビスマスを用いる。内標準液
の調製には,21.6.1のe)〜g) で規定する溶液のうち内標準とする元素の溶液を用いる。
e) カルシウム標準液(Ca:1 mg/L) c) のカルシウム標準液(Ca:1 000 mg/L)10 mLを全量フラスコ
100 mLにとり,水を標線まで加える。さらに,この溶液10 mLを全量フラスコ1 000 mLにとり,硝
酸(1+1)10 mLを加え,水を標線まで加える。使用時に調製する。
22.5.2
器具及び装置
器具及び装置は,次による。
a) ICP質量分析計 JIS K 0133による。ICP質量分析計は,カルシウム,鉄などの定量において,スペ
クトル干渉となるAr(40),ArO(56)などの分子イオンのピークによる干渉を低減できるものを用
いる。干渉を低減する方法としてプラズマの温度を低くする方式の装置がある。また,コリジョン・
リアクションセルなどのスペクトル干渉除去機能が搭載されている装置もある。
22.5.3
準備操作
準備操作は,次による。
a) 試料の適量を全量フラスコ100 mLにとり,内標準溶液(1 mg/L)1 mLを加え,硝酸の最終濃度が0.1
〜0.5 mol/Lになるように硝酸(1+1)を加え,水を標線まで加える。このときの濃度が表12の定量
範囲になるようにする。
b) 空試験の溶液として水を全量フラスコ100 mLにとり,a) と同じ内標準溶液(1 mg/L)1 mLを加え,
試料と同じ酸の濃度になるように硝酸(1+1)を加えた後,水を標線まで加える。
c) JIS K 0133の箇条8(分析装置の最適化),箇条9(分析条件の決定)に従って,ICP質量分析計を定
量できる状態に調整する。
22.5.4
操作
操作は,次による。
a) 22.5.3 a) の試料をJIS K 0133の箇条12(定量分析)に準じて試料導入部を通して高周波プラズマ中
に噴霧してカルシウムと内標準物質との質量/電荷数(m/z)における指示値(目的元素の質量/電荷
数におけるイオン電流又はその比例値)を読み取り,カルシウムの指示値と内標準物質の指示値との
比を求める。
111
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) 空試験として22.5.3 b) の溶液についてa) の操作を行ってカルシウムの指示値と内標準物質の指示値
との比を求め,試料について得たカルシウムと内標準物質との指示値の比を補正する。
c) あらかじめ,次によって作成した検量線からカルシウムの濃度を求め,試料中のカルシウムの濃度
(ng/L又はµg/L)を算出する。
1) カルシウム標準液(Ca:1 mg/L)0.05〜50 mLを用いて22.5.3 a) に準じて検量線用標準液を調製す
る。これらの溶液についてa) の操作を行う。
2) 別に,空試験としてこの操作に用いた水を全量フラスコ100 mLにとり,内標準溶液(1 mg/L)1 mL
を加え,22.5.3 a) の試料と同じ酸の濃度になるように硝酸(1+1)を加えた後,同じ水を標線まで
加え,b) の操作を行ってそれぞれの標準液について得た指示値の比を補正し,カルシウムの濃度に
対する指示値と内標準元素の指示値との比の関係線を作成する。検量線の作成は,試料測定時に行
う。
22.5.5
留意事項
21.6.5による。
22.6
イオンクロマトグラフ法
21.8による。
23
マグネシウム(Mg)
23.1
一般事項
マグネシウムの定量には,キレート滴定法,フレーム原子吸光法,ICP発光分光分析法,ICP質量分析
法又はイオンクロマトグラフ法を適用する。
23.2
キレート滴定法
試料に緩衝液を加えてpHを10に調節し,指示薬としてエリオクロムブラックT[3-ヒドロキシ-4-[(1-
ヒドロキシ-2-ナフタレニル)アゾ]-7-ニトロ-1-ナフタレンスルホン酸ナトリウム]を加え,エチレンジア
ミン四酢酸二水素二ナトリウム溶液で滴定し,カルシウムとマグネシウムの合量に対する滴定量を求め,
カルシウムに対する滴定量を差し引き,マグネシウムを定量する。
定量範囲:MgとCaとの合量がMgとして1 mg/L以上,繰返し精度:2〜10 %
10.2.1によって滴定された10 mmol/L EDTA溶液の量及び22.2によって滴定された10 mmol/L EDTA溶
液の量を用いて,次の式によってマグネシウム濃度を算出する。
0
243
.0
000
1
Ca
×
×
−
=
V
b
V
a
M
ここに,
M: マグネシウム濃度(mg/L)
a: 10.2.1で滴定に要した10 mmol/L EDTA溶液(mL)
b: 22.2で滴定に要した10 mmol/L EDTA溶液(mL)
V: 10.2.1での試料(mL)
VCa: 22.2での試料(mL)
0.243 0: 10 mmol/L EDTA溶液1 mLのマグネシウム相当量(mg)
23.3
フレーム原子吸光法
試料をアセチレン-空気フレーム中に噴霧し,マグネシウムによる原子吸光を波長285.2 nmで測定して
マグネシウムを定量する。
定量範囲:Mg:0.02〜0.4 mg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
23.3.1
試薬
112
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
試薬は,次による。
a) 水 21.2.1 a) による。
b) 塩酸(1+1) 21.5.1 b) による。
c) ランタン(III)溶液(50 g/L) 22.3.1 c) による。
d) マグネシウム標準液(Mg:1 000 mg/L) 21.5.1 e) による。
e) マグネシウム標準液(Mg:2 mg/L) マグネシウム標準液(Mg:1 000 mg/L)10 mLを全量フラスコ
100 mLにとり,水を標線まで加える。この溶液10 mLを全量フラスコ500 mLにとり,塩酸(1+1)
10 mLを加えた後,水を標線まで加える。
23.3.2
器具及び装置
器具及び装置は,次による。
a) フレーム原子吸光分析装置 JIS K 0121で規定するフレーム原子吸光分析装置で,測定対象元素用の
光源を備え,かつ,バックグラウンド補正が可能なもの。
23.3.3
準備操作
準備操作は,次による。
a) 懸濁物質が含まれる試料などでは,21.2.4 a) によって試料を処理した後,試料の適量を全量フラスコ
100 mLにとり,塩酸(1+1)2 mLを加えた後,水を標線まで加える。このときの濃度が20〜400 μg/L
の定量範囲になるようにする。
b) この溶液10 mLを乾いたビーカにとり,ランタン(III)溶液(50 g/L)1 mLを加える。
c) 空試験として試料と同量の水をとり,a)〜b) の操作を行う。
23.3.4
操作
操作は,次による。
a) 22.3.3 b) の溶液をJIS K 0121の8.(操作方法)の操作に従ってフレーム中に噴霧し,波長285.2 nm
の指示値(吸光度又はその比例値)を読み取る。
b) 空試験として23.3.3 c) についてa) の操作を行い,試料について得た指示値を補正する。
c) あらかじめ,次によって作成した検量線から試料中のマグネシウムの濃度(mg/L)を算出する。
1) マグネシウム標準液(Mg:2 mg/L)1〜20 mLを全量フラスコ100 mLに段階的にとり,試料と同じ
酸の濃度になるように塩酸(1+1)を加えた後,水を標線まで加える。これらの溶液について23.3.3
b) の操作を行って検量線用標準液を調製し,a) の操作を行う。
2) 別に,空試験としてこの操作に用いた水について試料と同じ酸の濃度となるように塩酸(1+1)を
加えた後,23.3.3 b) 操作を行った溶液についてb) の操作を行って,それぞれの標準液について得
た指示値を補正し,マグネシウム(Mg)の濃度と指示値との関係線を作成する。検量線の作成は試
料測定時に行う。
23.3.5
留意事項
a) アルミニウムは少量(2 mg/L)でも妨害するが,ランタン(III)溶液(50 g/L)の添加によって抑制
することができる。
23.4
ICP発光分光分析法
試料を試料導入部を通して誘導結合プラズマ中に噴霧し,マグネシウムによる発光強度を波長279.553
nmで測定して,マグネシウムを定量する。この方法によって,マグネシウムのほかに表9に示す元素が
同時定量できる。
23.4.1
試薬
113
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
試薬は,21.5.1による。
23.4.2
器具及び装置
器具及び装置は,21.5.2による。
23.4.3
準備操作
準備操作は,21.5.3による。
23.4.4
操作
操作は,21.5.4による。
23.4.5
留意事項
留意事項は,21.5.5による。
23.5
ICP質量分析法
21.6による。
23.6
イオンクロマトグラフ法
21.8による。
24
銅(Cu)
24.1
一般事項
銅の定量には,箇条6の操作を行った試料について,ジエチルジチオカルバミド酸吸光光度法,クプリ
ゾン吸光光度法,ジンコン吸光光度法,フレーム原子吸光法,電気加熱原子吸光法,ICP発光分光分析法
又はICP質量分析法を適用する。
24.2
ジエチルジチオカルバミド酸吸光光度法
試料中に共存する金属のマスキング剤として,くえん酸塩及びエチレンジアミン四酢酸二水素二ナトリ
ウム(EDTA)を加え,アンモニア水でpHを9とした後,N,N-ジエチルジチオカルバミド酸ナトリウム(ジ
エチルカルバモジチオ酸ナトリウム)を加え,生成する黄褐色の銅錯体を酢酸ブチルで抽出し,その吸光
度を測定して銅を定量する。
定量範囲:吸収セル10 mmの場合Cu:0.02〜0.3 mg/L,繰返し精度:2〜10 %
24.2.1
試薬
試薬は,次による。
a) 水 JIS K 0557で規定するA3の水。定量する元素について空試験を行って使用に支障のないことを
確認しておく。
b) アンモニア水(1+1) JIS K 8085で規定するアンモニア水を用いて調製する。
c) 硫酸ナトリウム JIS K 8987で規定するもの。
d) くえん酸水素二アンモニウム溶液(100 g/L) JIS K 8284で規定するくえん酸水素二アンモニウム10
gを水に溶かして100 mLとする。
くえん酸水素二アンモニウム中に銅が含まれるときの精製は,次による。
1) くえん酸水素二アンモニウム10 gを水80 mLに溶かし,アンモニア水(1+1)を加えてpHを約9
とした後,水を加えて100 mLとする。
2) これを分液漏斗に入れ,f) のジエチルジチオカルバミド酸ナトリウム溶液(10 g/L)2 mL及びh) の
酢酸ブチル10 mLを加えて,激しく振り混ぜて放置する。
3) 水層を乾いたろ紙でろ過し,酢酸ブチルの小滴を除いたろ液を用いる。
e) EDTA溶液 JIS K 8107で規定するエチレンジアミン四酢酸二水素二ナトリウム二水和物2 gを水に
114
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
溶かして100 mLとする。
f)
ジエチルジチオカルバミド酸ナトリウム溶液(10 g/L) JIS K 8454で規定するN,N-ジエチルジチオ
カルバミド酸ナトリウム三水和物1.3 gを水に溶かして100 mLとする。着色瓶に保存し,2週間以上
経過したものは使用しない。
g) メタクレゾールパープル溶液(1 g/L) JIS K 8889で規定するメタクレゾールパープル0.1 gをJIS K
8102で規定するエタノール(95)50 mLに溶かし,水を加えて100 mLとする。
h) 酢酸ブチル JIS K 8377で規定するもの。
i)
銅標準液(Cu:100 mg/L) 国家計量標準にトレーサブルな銅標準液(Cu:100 mg/L)を使用するか,
又は,次による。JIS K 8005で規定する容量分析用標準物質の銅を塩酸(1+3)で洗い,水洗し,JIS
K 8101で規定するエタノール(99.5)で洗い,次に,JIS K 8103で規定するジエチルエーテルで洗っ
た後,直ちにデシケータ中に入れ,12時間以上放置する。Cu 100 %に対してその0.100 gをとり,硝
酸(1+1)(JIS K 8541で規定する硝酸を用いて調製する。)20 mL中に加え,煮沸して溶かし,窒素
酸化物を追い出す。放冷後,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。または,JIS K
8983で規定する硫酸銅(II)五水和物0.393 gをとり,硝酸(1+1)20 mLを加えて溶かし,全量フラ
スコ1 000 mLに移し入れ,水を標線まで加える。
j)
銅標準液(Cu:1 mg/L) 銅標準液(Cu:100 mg/L)10 mLを全量フラスコ1 000 mLにとり,硝酸
(1+1)20 mLを加え,水を標線まで加える。
24.2.2
器具及び装置
器具及び装置は,次による。
a) 分液漏斗 100 mL又は300 mL
b) 光度計 分光光度計又は光電光度計
24.2.3
準備操作
準備操作は,次による。
a) 試料100 mLについて箇条6の操作を行い,液量を約30 mL以下にする。
b) 有機物及び濁りを含まない試料で銅の濃度が低い場合には,試料250 mLまでの適量をとり,箇条6
の前処理で約30 mL以下に濃縮をする。
c) 試料の銅濃度が0.3 mg/L以上の場合は,試料量を減らして操作を行う。
24.2.4
操作
操作は,次による。
a) 24.2.3で前処理した試料を分液漏斗にとり,指示薬としてメタクレゾールパープル溶液(1 g/L)2,3
滴を加えた後,くえん酸水素二アンモニウム溶液(100 g/L)5 mL及びEDTA溶液1 mLを加える。
b) アンモニア水(1+1)を加えて溶液の色が僅かに紫を呈するまで(pH約9)中和し,水を加えて50 mL
とする。
c) ジエチルジチオカルバミド酸ナトリウム溶液(10 g/L)2 mLを加えて混合し,次に,酢酸ブチル10 mL
を加え,約3分間激しく振り混ぜて,放置する。
d) 水層を捨て,酢酸ブチル層を硫酸ナトリウム約1 gを入れた共栓試験管に移し,振り混ぜる。試験管
に移す場合は,乾いたろ紙でろ過するか,又は分液漏斗の脚部に乾いた脱脂綿を詰めてろ過する。
e) その一部を吸収セルに移し,酢酸ブチルを対照液として波長440 nm付近の吸光度を測定する。
f)
空試験として試料と同量の水をとり,試料に適用した箇条6の前処理を行って空試験の溶液とし,a)
の試料と同量をとり,a)〜e) の操作を行って吸光度を求め,試料について得た吸光度を補正する。
115
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
g) あらかじめ,次によって作成した検量線から銅の濃度を求め,試料中の銅の濃度(mg/L)を算出する。
1) 銅標準液(Cu:1 mg/L)2〜30 mLを分液漏斗に段階的にとり,a)〜f) の操作を行って銅(Cu)の
濃度と吸光度との関係線を作成する。
24.2.5
留意事項
a) 試料中に陰イオン界面活性剤(例えば,スルホン酸形のもの),タンニンなどが含まれるときは,銅の
抽出が不完全になる。
b) ビスマスは,銅とともに抽出され黄色を示すが,銅の量の2倍量以下のときは,ほとんど影響しない。
2倍量以上を含むときは,24.2.4の操作に従って測定した吸光度をA1とし,別に,銅の試験に用いた
試料と同量の試料をとり,操作24.2.4 a) の後に,シアン化カリウム溶液(50 g/L)3 mLを加え,銅を
シアノ錯体とした後,24.2.4のb)〜f) の操作を行ってビスマス錯体だけを抽出し,その吸光度をA2
とする。銅による吸光度はA1−A2である。
c) EDTA溶液を加えない場合は,ジエチルジチオカルバミド酸ナトリウムは多くの金属元素と反応する。
しかし,水銀,ひ素,鉛,すず,アンチモンなど,大部分の金属錯体は無色である。鉄,ニッケル,
コバルトなどの錯体は,有色であるが,この方法では,EDTA溶液によってマスキングされる。
24.3
クプリゾン吸光光度法
この方法は,銅(II)がpH 8.5〜9.2でクプリゾン(ビスシクロヘキサノンオキサリルジヒドラゾン)と
反応して生じる錯体の青い色の吸光度を測定して銅を定量する方法である。
定量範囲:吸収セル50 mmの場合Cu:0.01〜0.2 mg/L,吸収セル20 mmの場合Cu:0.025〜0.5 mg/L,
繰返し精度:2〜10 %
24.3.1
試薬
試薬は,次による。
a) 水 24.2.1 a) による。
b) くえん酸水素二アンモニウム溶液(200 g/L) JIS K 8284で規定するくえん酸水素二アンモニウム100
gを水に溶かして約400 mLとし,アンモニア水(1+1)を加えてpHを約8.5とした後,水を加えて
500 mLとする。
c) アンモニア水(1+1) 24.2.1 b) による。
d) クプリゾン溶液(1 g/L) クプリゾン(ビスシクロヘキサノンオキサリルジヒドラゾン)0.5 gをとり,
JIS K 8102で規定するエタノール(95)50 mLを加え,沸騰水浴上で加熱して溶かす。不溶解物があ
るときはろ別し,水を加えて500 mLとする。
e) 銅標準液(Cu:10 mg/L) 銅標準液(Cu:100 mg/L)10 mLを全量フラスコ100 mLにとり,硝酸(1
+1)2 mLを加え,水を標線まで加える。
24.3.2
器具及び装置
器具及び装置は,次による。
a) 光度計 分光光度計又は光電光度計
b) メスシリンダ(有栓形) 15.2.2 b) による。
24.3.3
準備操作
準備操作は,次による。
a) 試料100 mLについて箇条6の操作を行い,液量を約30 mL以下にする。
b) 試料の銅濃度が0.5 mg/L以上の場合は,試料量を減らして操作を行う。
24.3.4
操作
116
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
操作は,次による。
a) 24.3.3で前処理した試料の全量をビーカにとる。
b) くえん酸水素二アンモニウム溶液(200 g/L)5 mLを加えて振り混ぜる。
c) クプリゾン溶液(1 g/L)10 mLを加え,アンモニア水(1+1)を滴加してpHを8.5〜9.2[pH試験紙
チモールブルー(pH 8.0〜9.6)又はメタクレゾールパープル(pH 7.4〜9.0)を用いる。]に調節する。
液温を25 ℃以下(液温が高いときは,呈色が不安定になる。)に冷却する。
d) メスシリンダ(有栓形)50 mLに移し入れ,水を50 mLの標線まで加えて振り混ぜ,約5分間放置す
る。
e) この溶液の一部を吸収セル50 mmに移し,波長600 nm付近の吸光度を測定する。
f)
別に,空試験として試料と同量の水をとり,試料に適用した箇条6の操作を行って空試験用の溶液と
し,b)〜e) の操作を行って試料について得た吸光度を補正する。
g) あらかじめ,次によって作成した検量線から銅の濃度を求め,試料中の銅の濃度(mg/L)を算出する。
1) 銅標準液(Cu:10 mg/L)0.1〜2 mLをビーカに段階的にとり,b)〜c) の操作を行う。e) で20 mm
の吸収セルを使用する場合は,銅標準液(Cu:10 mg/L)0.25〜5 mLをビーカに段階的にとり,b)
〜c) の操作を行う。
2) 別に,空試験として水について同様の操作を行ってそれぞれの標準液について得た吸光度を補正し,
銅(Cu)の濃度と吸光度との関係線を作成する。
24.3.5
留意事項
a) クロム(III)0.5 mg,ニッケル4 mg,コバルト0.2 mg,マンガン(II)5 mg,バナジウム4 mg及びチ
タン7.5 mg以上が共存すると妨害する。すず及び鉛は酒石酸アンモニウム溶液を添加すれば,妨害を
除去できる。
b) くえん酸水素二アンモニウム溶液(200 g/L)に代えて,酒石酸アンモニウム溶液(100 g/L)[(+)-酒
石酸アンモニウム10 gを水に溶かして100 mLとする。]10 mLを加えると,すず10 mg,鉛100 mg
までの妨害を除去できる。
24.4
ジンコン吸光光度法
この方法は,試料中に共存する金属元素のマスキング剤として酒石酸を加え,酢酸アンモニウム溶液を
加えてpH約4でジンコン[2-[1-(2-ヒドロキシ-5-スルホフェニル)-3-フェニル-5-ホルマザノ]安息香
酸一ナトリウム]と反応して生じる錯体の青い色の吸光度を測定して銅を定量する。
定量範囲:吸収セル50 mmの場合Cu:5〜100 μg/L,繰返し精度:2〜10 %
24.4.1
試薬
試薬は,次による。
a) 水 JIS K 0557で規定するA3又はA4の水。定量する元素について空試験を行って使用に支障のない
ことを確認しておく。
b) 酒石酸溶液(1 mol/L) JIS K 8532で規定するL(+)-酒石酸15 gを水に溶かして100 mLとする。
c) 酢酸アンモニウム溶液(500 g/L) JIS K 8359で規定する酢酸アンモニウム500 gを水に溶かして1 L
とする。酢酸アンモニウム溶液(500 g/L)中に銅が含まれるときの精製は,次による。
1) 酢酸アンモニウム溶液(500 g/L)100 mLを分液漏斗に入れ,ジンコン・3-メチル-1-ブタノール溶液
(ジンコン溶液2 mLをJIS K 8051で規定する3-メチル-1-ブタノール100 mLに溶かす。)20 mLを
加えてよく振り混ぜ放置する。
2) 分離した有機溶媒層を捨てる。
117
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3) この操作を銅による着色が認められなくなるまで繰り返す。
d) ジンコン溶液 ジンコン“o-[2-[α-(2-ヒドロキシ-5-スルホフェニルアゾ)-ベンジリデン]-ヒドラ
ジノ]安息香酸一ナトリウム”75 mgをとり,JIS K 8891で規定するメタノール[又はJIS K 8102で
規定するエタノール(95)を用いてもよい。]50 mLを加えて加熱(約50 ℃)して溶かし,水で100 mL
とし,着色瓶に入れる。使用時に調製する。
e) 銅標準液(Cu:1 mg/L) 24.2.1 j) による。
24.4.2
器具及び装置
器具及び装置は,次による。
a) 光度計 分光光度計又は光電光度計
b) メスシリンダ(有栓形) 15.2.2 b) による。
24.4.3
準備操作
準備操作は,次による。
a) 試料100 mLについて箇条6の操作を行い,液量を約30 mL以下にする。
b) 試料の銅濃度が100 μg/L以上の場合は,試料量を減らして操作を行う。
24.4.4
操作
操作は,次による。
a) 24.4.3で前処理した試料の全量を,メスシリンダ(有栓形)50 mLにとる。
b) 酒石酸溶液(1 mol/L)2 mLを加え,酢酸アンモニウム溶液(500 g/L)を加えて,pHを3.8〜4.8に調
節する。
c) ジンコン溶液0.1 mLを加え,水を50 mLの標線まで加えて振り混ぜる。
d) 別に,空試験として試料と同量の水について,試料に適用した箇条6の操作を行って空試験用の溶液
とし,a) と同量をメスシリンダ(有栓形)50 mLにとり,b)〜c) の操作を行う。
e) 試料についてc) で得た溶液の一部を吸収セルに移し,d) で得た溶液を対照液として,波長600 nm付
近の吸光度を測定する。
f)
あらかじめ,次によって作成した検量線から銅の濃度を求め,試料中の銅の濃度(mg/L)を算出する。
1) 銅標準液(Cu:1 mg/L)0.5〜10 mLを段階的にメスシリンダ(有栓形)50 mLにとり,JIS K 8180
で規定する塩酸4 mLを加え,水を加えてb)〜c) の操作を行う。
2) 別に,水について同様の操作を行ってこの溶液を対照液としてそれぞれの標準液の吸光度を測定し,
銅(Cu)の濃度と吸光度との関係線を作成する。検量線は測定時に作成する。
24.4.5
留意事項
a) この方法では,pH 3以上でジンコン銅錯体の吸光度は一定となるが,ニッケルの妨害を考慮すると,
pH 3.5〜4.8が望ましい。操作では,pHを3.8〜4.3になるように調節している。また,酒石酸は鉄(II)
の妨害をマスキングしている。
24.5
フレーム原子吸光法
試料を前処理した後,アセチレン-空気フレーム中に噴霧し,銅による原子吸光を波長324.8 nmで測定
して銅を定量する。
定量範囲:Cu:0.2〜4 mg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
24.5.1
試薬
試薬は,次による。
a) 水 24.2.1 a) による。
118
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) 硝酸(1+1) JIS K 8541で規定する硝酸を用いて調製する。
c) 銅標準液(Cu:10 mg/L) 24.2.1 i) の銅標準液(Cu:100 mg/L)50 mLを全量フラスコ500 mLにと
り,硝酸(1+1)10 mLを加えた後,水を標線まで加える。
24.5.2
器具及び装置
器具及び装置は,次による。
a) フレーム原子吸光分析装置 JIS K 0121で規定するフレーム原子吸光分析装置で,測定対象元素用の
光源を備え,かつ,バックグラウンド補正が可能なもの。
24.5.3
準備操作
準備操作は,次による。
a) 試料を6.6によって前処理する。
b) 空試験としてa) の準備操作での試料と同量の水をとり,試料と同様にa) の操作を行う。
c) 試料中のナトリウム,カリウム,カルシウム,マグネシウムなどの濃度が高く,銅の濃度が低い試料
で,抽出操作を妨害する物質を含まない場合の準備操作は,次による。
1) 試料500 mL(又は100〜500 mLの一定量)をビーカにとり,JIS K 8180で規定する塩酸10 mLを
加え,約5分間煮沸する。
2) 放冷後,分液漏斗1 000 mL(又は200〜500 mL)に移し入れ,くえん酸水素二アンモニウム溶液(100
g/L)[24.2.1 d) による。]10 mL及び指示薬としてメタクレゾールパープル溶液(1 g/L)[24.2.1 g) に
よる。]2,3滴を加えた後,アンモニア水(1+1)を溶液の色が僅かに紫に呈色するまで加える。
3) ジエチルジチオカルバミド酸ナトリウム溶液(10 g/L)[24.2.1 f) による。]5 mLを加えて振り混ぜ
た後,JIS K 8377で規定する酢酸ブチル20 mLを加え,約1分間激しく振り混ぜ,静置する。
4) 酢酸ブチル層を分離し,ビーカ100 mLに入れる。水層に酢酸ブチル5 mLを加えて抽出操作を繰り
返す。抽出した酢酸ブチル層は先のビーカに合わせる。
5) 加熱して酢酸ブチルを揮散させた後,JIS K 8541で規定する硝酸2 mLとJIS K 8223で規定する過
塩素酸2 mLを加えて加熱し,有機物を分解する。ほとんど乾固した後,放冷する。
6) 残留物を硝酸(1+15)(JIS K 8541で規定する硝酸を用いて調製する。)10 mLに溶かし,これを銅
の定量に用いる(亜鉛,ニッケルなどの定量に使用できる。)。
7) 抽出した酢酸ブチル層に酢酸ブチルを加えて液量を一定量にしたもの,又は抽出条件を一定にして,
1回抽出を行った酢酸ブチル層をそのまま噴霧して原子吸光分析することもできる。
d) c) のほかの準備操作は,次による。
1) 試料200 mLをとり,6.6と同様に酸処理し,pHを3.5〜4.0に調節する。
2) 硫酸アンモニウム溶液(飽和)20 mLを加える。1-ピロリジンカルボジチオ酸アンモニウム[ピロ
リジン-N-ジチオカルバミド酸アンモニウム(APDC)]溶液(10 g/L)5 mLを加え,静かに振りま
ぜた後,約3分間放置する。
3) JIS K 8903で規定する4-メチル-2-ペンタノン10 mLを加え,約3分間激しく振り混ぜ,静置する。
4) 有機層を分離し,ビーカ100 mLに入れる。水層に4-メチル-2-ペンタノン5 mLを加え,抽出操作を
繰り返す。抽出した有機層は先のビーカに合わせる。
5) この有機層を加熱して4-メチル-2-ペンタノンを揮散させた後,JIS K 8541で規定する硝酸2 mLと
JIS K 8223で規定する過塩素酸2 mLを加えて加熱し,有機物を分解する。ほとんど乾固した後,
放冷する。
6) 残留物を硝酸(1+15)10 mLに溶かし,これを銅の定量に用いる(亜鉛,鉛,カドミウム,ニッケ
119
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ルなどの定量に使用できる。)。
7) 抽出した4-メチル-2-ペンタノン層に同じ溶媒を加えて一定量としたもの,又は抽出条件を一定にし
て,1回抽出を行った4-メチル-2-ペンタノン層をそのまま噴霧して原子吸光分析することもできる。
24.5.4
操作
操作は,次による。
a) 24.5.3 a) の準備操作を行った試料を,JIS K 0121の8.(操作方法)の操作に従ってフレーム中に噴霧
し,波長324.8 nmの指示値(吸光度又はその指示値)を読み取る。
b) 空試験として24.5.3 b) の準備操作の水を試料と同様にa) の操作を行って試料について得た指示値を
補正する。
c) あらかじめ,次によって作成した検量線から銅の濃度を求め,試料中の銅の濃度(mg/L)を算出する。
1) 銅標準液(Cu:10 mg/L)2〜40 mLを全量フラスコ100 mLに段階的にとり,24.5.3 a) を行った試
料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。これらの溶液についてa) の
操作を行う。
2) 別に,空試験としてこの操作に用いた水について24.5.3 a) を行った試料と同じ酸の濃度になるよう
に酸を加えた後,a) の操作を行ってそれぞれの標準液について得た指示値を補正し,銅(Cu)の濃
度と指示値との関係線を作成する。検量線の作成は,試料測定時に行う。
3) 準備操作として溶媒抽出を適用するときは,銅標準液(Cu:10 mg/L)の量を,適宜,減らす。24.5.3
b) 及びc) の抽出によって準備操作を行い,酢酸ブチル層,4-メチル-2-ペンタノン層をそのまま噴
霧する場合の検量線は,銅標準液(Cu:10 mg/L)を適切な濃度(Cu:0.1〜1 mg/L)に薄め,その
2〜40 mLを段階的にとり,500 mL(又は100〜500 mLの一定量)とした後,試料と同様にa) 及び
b) を行って銅(Cu)の濃度と指示値との関係線を作成する。
24.5.5
留意事項
a) 24.5.3 c) の酢酸ブチルに代え,JIS K 8903で規定する4-メチル-2-ペンタノン又は2,6-ジメチル-4-ヘプ
タノン(ジイソブチルケトン,DIBK)を用いてもよい。2,6-ジメチル-4-ヘプタノンを用いる場合は,
2,6-ジメチル-4-ヘプタノンは水との相互溶解がほとんどないので,その添加量は少なくてもよい。
b) 24.5.3 d) の4-メチル-2-ペンタノンの代わりに2,6-ジメチル-4-ヘプタノンを用いてもよい。
24.6
電気加熱原子吸光法
試料を前処理した後,電気加熱炉で原子化し,銅による原子吸光を波長324.8 nmで測定して銅を定量す
る。
定量範囲:Cu:5〜100 µg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
この方法は,共存する酸,塩の種類及び濃度の影響を受けやすいので,これらの影響の少ない試料(例
えば,貫流ボイラの給水,揮発性物質処理を行っている水管ボイラの給水及びボイラ水の試料。)に適用す
る。
24.6.1
試薬
試薬は,次による。
a) 水 24.4.1 a) による。
b) 硝酸(1+1) 21.6.1 b) による。
c) 銅標準液(Cu:1 mg/L) 24.2.1 j) による。
24.6.2
器具及び装置
器具及び装置は,次による。
120
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) 電気加熱原子吸光分析装置 JIS K 0121で規定する電気加熱原子吸光分析装置で,測定対象元素用の
光源を備え,かつ,バックグランド補正が可能なもの。
b) マイクロピペット JIS K 0970で規定するピストン式ピペット10〜50 µL,又は自動注入装置
24.6.3
準備操作
準備操作は,次による。
a) 試料を6.6によって前処理する。
b) 空試験としてa) の準備操作での試料と同量の水をとり,試料と同様にa) の操作を行う。
24.6.4
操作
操作は,次による。
a) 24.6.3 a) の準備操作を行った試料の一定量(例えば,10〜50 µL)をマイクロピペットで発熱体に注
入し,JIS K 0121の8.(操作方法)の操作に準じて,乾燥(100〜120 ℃,30〜40秒間)した後,灰
化(600〜1 000 ℃,30〜40秒間)し,次に,原子化(2 200〜2 700 ℃,3〜6秒間)し,波長324.8 nm
の指示値(吸光度又はその指示値)を読み取る。この操作を3回繰り返し,指示値が合うことを確認
する。
なお,乾燥,灰化,原子化の温度と時間は用いる装置,試料の注入量及び共存する塩類の濃度によ
って異なるので,装置及び試料によってこれらの条件の最適化をはかる。
b) 空試験として24.6.3 b) の準備操作の水を試料と同様にa) の操作を行って試料について得た指示値を
補正する。
c) あらかじめ,次によって作成した検量線から銅の濃度を求め,試料中の銅の濃度(µg/L)を算出する。
1) 銅標準液(Cu:1 mg/L)0.5〜10 mLを全量フラスコ100 mLに段階的にとり,24.6.3 a) を行った試
料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。この溶液についてa) の操作
を行う。
2) 別に,空試験として水について24.6.3 a) を行った試料と同じ酸の濃度となるように酸を加えた後,
a) の操作を行って標準液について得た指示値を補正し,銅(Cu)の濃度と指示値との関係線を作成
する。検量線の作成は,試料測定時に行う。
24.6.5
留意事項
a) 試料中の銅濃度が極低濃度の場合には,試料の一定量(10〜50 μL)を発熱体に注入し,乾燥する。こ
の操作を繰り返した後,灰化,原子化の操作を行って指示値を読み取る。この場合は,24.6.4 b) の操
作でも同様な回数を注入し,乾燥する。
24.7
ICP発光分光分析法
試料を前処理した後,試料導入部を通して誘導結合プラズマ中に噴霧し,銅による発光を波長324.754 nm
で測定して銅を定量する。この方法によって表13に示す元素が同時定量できる。それぞれの元素ごとの
測定波長,定量範囲及び繰返し精度の例を,表13に示す。

121
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表13−測定波長,定量範囲及び繰返し精度の例a)
対象元素
測定波長
nm
定量範囲
繰返し精度
μg/L
%
銅(Cu)
324.754
50〜5 000
2〜10
亜鉛(Zn)
213.856
10〜6 000
2〜10
ニッケル(Ni)
221.647
40〜2 000
2〜10
鉄(Fe)
238.204
10〜5 000
2〜10
イットリウム(Y)b)
371.029
−
−
注a) 装置及び測光方式,測定条件によって異なる。
b) 内標準元素として,イットリウム(Y)のほか,インジウム(In)及びイッテルビウム(Yb)
も使用できる。
24.7.1
試薬
試薬は,次による。
a) 水 24.4.1 a) による。
b) 硝酸(1+1) 21.6.1 b) による。
c) 銅標準液(Cu:100 mg/L) 24.2.1 i) による。
d) 亜鉛標準液(Zn:100 mg/L) 国家計量標準にトレーサブルな亜鉛標準液(Zn:100 mg/L)を使用す
るか,又は,次による。JIS K 8005で規定する容量分析用標準物質の亜鉛を塩酸(1+3)で水洗いし,
JIS K 8101で規定するエタノール(99.5)で洗い,次に,JIS K 8103で規定するジエチルエーテルで
洗った後,直ちに上口デシケータ中に入れ,圧力2 kPa以下で数分間保った後,減圧化で約12時間保
つ。Zn 100 %に対してその0.100 gをとり,硝酸(1+1)30 mLに溶かし,沸騰して窒素酸化物を追い
出す。放冷後,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
e) ニッケル標準液(Ni:100 mg/L) 国家計量標準にトレーサブルなニッケル標準液(Ni:100 mg/L)
を使用するか,又は,次による。JIS K 9062で規定するニッケル(99.9 %以上)0.100 gをとり,硝酸
(1+1)20 mL中に加え,煮沸して溶かし,窒素酸化物を追い出す。放冷後,全量フラスコ1 000 mL
に移し入れ,水を標線まで加える。又は,硫酸ニッケル(II)アンモニウム六水和物0.673 gをとり,
水と硝酸(1+1)10 mLとに溶かし,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
f)
鉄標準液(Fe:1 000 mg/L) 国家計量標準にトレーサブルな鉄標準液(Fe:1 000 mg/L)を使用する
か,又は,次による。鉄(99.5 %以上)1.000 gをとり,塩酸(1+1)(JIS K 9902で規定する高純度
試薬−塩酸で調製する。)30 mL中に入れ加熱して溶かし,放冷後,全量フラスコ1 000 mLに移し入
れ,水を標線まで加える。又は,JIS K 8979で規定する硫酸アンモニウム鉄(II)六水和物7.02 gを
とり,塩酸(1+1)20 mLと少量の水とに溶かし,全量フラスコ1 000 mLに移し入れ,水を標線まで
加える。
g) 鉄標準液(Fe:100 mg/L) 鉄標準液(Fe:1 000 mg/L)10 mLを全量フラスコ100 mLにとり,硝酸
(1+1)2 mLを加えた後,水を標線まで加える。
h) イットリウム溶液(Y:1 000 mg/L) 21.5.1 f) による。
i)
イットリウム溶液(Y:50 mg/L) 21.5.1 g) による。
j)
混合標準液[(Cu:10 mg,Zn:10 mg,Ni:10 mg,Fe:10 mg)/L] 銅標準液(Cu:100 mg/L)10 mL,
d) の亜鉛標準液(Zn:100 mg/L)10 mL,e) のニッケル標準液(Ni:100 mg/L)10 mL,g) の鉄標準
液(Fe:100 mg/L)10 mLをそれぞれ全量フラスコ100 mLにとり,硝酸(1+1)2 mLを加え,水を
122
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
標線まで加える。使用時に調製する。
24.7.2
器具及び装置
器具及び装置は,次による。
a) ICP発光分光分析装置 JIS K 0116による。
24.7.3
準備操作
準備操作は,次による。
a) 試料を6.6によって前処理する。
b) 空試験として,試料と同量の水を用いてa) の操作を行う。
24.7.4
操作
操作は,次による。
a) 24.7.3 a) の溶液をJIS K 0116の4.7(定量分析)に準じて,試料導入部を通してプラズマ中に噴霧し,
波長324.754 nmの発光強度を測定する。
b) 空試験として24.7.3 b) についてa) の操作を行って,試料について得た発光強度を補正する。
c) 検量線から銅の濃度を求め,試料中の銅の濃度(mg/L又はμg/L)を算出する。
d) 銅と同時に亜鉛,ニッケル,鉄を同時定量する場合には,a)〜c) においてそれぞれの元素における発
光強度を測定し,銅の場合と同様にそれぞれの金属元素について作成した検量線を用いて,それぞれ
の元素の濃度を算出する。
1) 混合標準液[(Cu:10 mg,Zn:10 mg,Ni:10 mg,Fe:10 mg)/L]を全量フラスコ100 mLに測定
濃度範囲を含むように,混合標準液を段階的にとり,24.7.3 a) の試料と同じ酸の濃度となるように
硝酸(1+1)を加えた後,水を標線まで加える。これらの溶液についてa) の操作を行う。
2) 別に,空試験としてこの操作に用いた水について試料と同じ条件になるように硝酸(1+1)を加え
た後,b) の操作を行ってそれぞれの標準液について得た発光強度を補正し,測定対象元素の濃度と
発光強度との関係線を作成する。検量線の作成は試料測定時に行う。
24.7.5
留意事項
a) 波長の異なる2本以上のスペクトル線を同時測定が可能な装置では,内標準法によることができる。
1) 内標準法を用いるときは,24.7.3 a) で処理した試料の適量を全量フラスコ100 mLにとり,イット
リウム溶液(Y:50 mg/L)10 mLを加え,24.7.3 a) の試料と同じ酸の濃度になるように酸を加えた
後,水を標線まで加える。
2) この溶液について24.7.4 a) の操作を行って測定対象元素の波長と同時に371.029 nm(イットリウ
ム)の発光強度を測定し,測定対象元素とイットリウムとの発光強度の比を求める。
3) 空試験として試料と同量の水を用いて,1) 及び2) の操作を行う。
4) あらかじめ,次によって作成した各測定対象元素の検量線から,試料中の測定対象元素の濃度を算
出する。
4.1) 混合標準液[(Cu:10 mg,Zn:10 mg,Ni:10 mg,Fe:10 mg)/L]を全量フラスコ100 mLに測定
濃度範囲を含むように,混合標準液を段階的にとり,21.5.1 g) のイットリウム溶液(Y:50 mg/L)
10 mLをそれぞれ加え,24.7.3 a) の試料と同じ酸の濃度になるように酸を加えた後,水を標線ま
で加える。これらの溶液について24.7.4 a) の噴霧操作を行って測定対象元素の各濃度における測
定対象元素とイットリウムとの発光強度比を求める。
4.2) 別に,空試験として混合標準液に代えて水を用い,同様に操作・測定を行って測定対象元素とイ
ットリウムとの発光強度比を求め,測定対象元素の発光強度比を補正する。各測定対象元素の濃

123
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
度と発光強度比の関係線を作成し,検量線とする。検量線の作成は試料測定時に行う。
b) ナトリウム,カリウム,マグネシウム,カルシウムなどの濃度が高く,銅の濃度が低い場合には,次
のように操作する。
1) 試料500 mL(又は100〜500 mLの一定量)をビーカにとり,JIS K 8180で規定する塩酸5 mLを加
え,約5分間煮沸する。
2) 放冷後,酢酸-酢酸ナトリウム緩衝液(pH 5)[JIS K 8371で規定する酢酸ナトリウム三水和物19.2 g
とJIS K 8355で規定する酢酸3.4 mLとを水に溶かして1 Lとする。]10 mLを加え,アンモニア水
(1+1)又は硝酸(1+10)でpHを5.2に調節する。
3) この溶液を分液漏斗1 000 mL(又は200〜500 mL)に移し,1-ピロリジンカルボジチオ酸アンモニ
ウム溶液(20 g/L)2 mL,ヘキサメチレンアンモニウム-ヘキサメチレンカルバモジチオ酸(ヘキサ
メチレンアンモニウム-ヘキサメチレンジチオカルバミド酸)のメタノール溶液(20 g/L)2 mLを加
えて混合した後,JIS K 8271で規定するキシレンの一定量(例えば,5〜20 mL)を加えて約5分間
激しく振り混ぜて静置する。
4) 水層を捨てキシレン層を共栓試験管に入れる。
5) キシレン層をそのまま噴霧する場合の検量線は混合標準液[(Cu:10 mg,Zn:10 mg,Ni:10 mg,
Fe:10 mg)/L]を適切な濃度に薄め,その0.2〜50 mLを段階的にとり,500 mL(又は100〜500 mL
の一定量)とした後,試料と同様に24.7.5の留意事項及びa) とb) の操作を行って測定対象元素の
濃度と発光強度との関係線を作成する。
注意 この操作に用いる酢酸-酢酸ナトリウム緩衝液(pH 5)は,使用前に1-ピロリジンカルボジチ
オ酸アンモニウム溶液,ヘキサメチレンアンモニウムヘキサメチレンカルバモジチオ酸のメ
タノール溶液及びキシレンを加えて振り混ぜ,精製したものを用いる。
24.8
ICP質量分析法
試料を前処理した後,内標準元素を加え,試料導入部を通して高周波プラズマ中に噴霧し,銅及び内標
準元素(イットリウム,インジウムマ又はビスマス)のそれぞれの質量/電荷数(m/z)における指示値(イ
オンカウント値又はその比例値)を測定し,銅の指示値と内標準元素の指示値との比を求めて銅を定量す
る。この方法によって,銅のほかに表14に示す元素が同時定量できる。それぞれの元素ごとの定量範囲,
繰返し精度などの例を,表14に示す。
表14−定量範囲,繰返し精度及び質量数の例a)
対象元素
定量範囲
繰返し精度
質量数
μg/L
%
銅(Cu)
0.5〜500
2〜10
63,65
亜鉛(Zn)
0.5〜500
2〜10
64,66,68
イットリウム(Y)b)
−
−
89
インジウム(In)b)
−
−
115
ビスマス(Bi)b)
−
−
209
注a) 装置及び測定条件によって異なる。
b) 内標準元素はいずれかを使用する。
24.8.1
試薬
試薬は,次による。
124
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) 水 24.4.1 a) による。
b) 硝酸(1+1) 21.6.1 b) による。
c) 塩酸(1+3) JIS K 8180で規定する塩酸を用いて調製する。
d) 銅標準液(Cu:100 mg/L) 24.2.1 i) による。
e) 亜鉛標準液(Zn:100 mg/L) 24.7.1 d) による。
f)
イットリウム溶液(Y:1 mg/L) 21.6.1 e) による。
g) インジウム溶液(In:1 mg/L) 21.6.1 f) による。
h) ビスマス溶液(Bi:1 mg/L) 21.6.1 g) による。
i)
混合標準液[(Cu:1 mg,Zn:1 mg)/L] d) の銅標準液(Cu:100 mg/L)10 mL,e) の亜鉛標準液
(Zn:100 mg/L)10 mLをそれぞれ全量フラスコ1 000 mLにとり,硝酸(1+1)1.5 mLを加え,水を
標線まで加える。使用時に調製する。ただし,単独の標準液を調製して使用してもよい。
24.8.2
器具及び装置
器具及び装置は,次による。
a) ICP質量分析計 JIS K 0133による。分子イオンのピークによる干渉を低減する方法としてプラズマ
の温度を低くする方式の装置がある。また,コリジョン・リアクションセルなどのスペクトル干渉除
去機能が搭載されている装置もある。この装置では,26.7に示す分析成分と同時測定を行うことがで
きる。
24.8.3
準備操作
準備操作は,次による。
a) 試料を6.6によって前処理し,全量フラスコ100 mLに移し入れ,いずれかの内標準溶液(1 mg/L)1 mL
を加え,硝酸の最終濃度が0.1〜0.5 mol/Lになるように硝酸(1+1)を加えた後,水を標線まで加え
る。このときの濃度が表14の定量範囲になるようにする。
b) 空試験の溶液として同量の水を6.6によって前処理し,全量フラスコ100 mLに移し入れ,a) と同じ
内標準溶液(1 mg/L)1 mLを加え,試料と同じ酸の濃度になるように硝酸(1+1)を加えた後,水を
標線まで加える。
c) JIS K 0133の箇条8(分析装置の最適化),箇条9(分析条件の決定)に準じて,ICP質量分析計を定
量できる状態に調整する。
24.8.4
操作
操作は,次による。
a) 24.8.3 a) の試料をJIS K 0133の箇条12(定量分析)に準じて,試料導入部を通して高周波プラズマ
中に噴霧して測定対象元素と内標準物質の質量/電荷数(m/z)における指示値[目的元素の(質量/
電荷数)におけるイオン電流又はその比例値]を読み取り,測定対象元素の指示値と内標準物質の指
示値との比を求める。
b) 空試験として24.8.3 b) の溶液についてa) の操作を行って測定対象元素の指示値と内標準物質の指示
値との比を求め,試料について得た測定対象元素と内標準物質との指示値の比を補正する。
c) 検量線から測定対象元素の濃度を求め,試料中の測定対象元素の濃度(ng/L又はµg/L)を算出する。
d) 銅と亜鉛とを同時定量する場合には,各金属元素について,それぞれの検量線を作成する。検量線の
作成は,試料測定時に行う。
1) 混合標準液[(Cu:1 mg,Zn:1 mg)/L]0.05〜50 mLを全量フラスコ100 mLに段階的にとり,内標
準溶液(1 mg/L)1 mLを加え,24.8.3 a) の試料と同じ酸の濃度になるように硝酸(1+1)を加え,
125
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
水を標線まで加える。これらの標準液についてa) の操作を行う。
2) 別に,空試験としてこの操作に用いた水を全量フラスコ100 mLにとり,内標準溶液(1 mg/L)1 mL
を加え,24.8.3 a) の試料と同じ酸の濃度になるように硝酸(1+1)を加えた後,同じ水を標線まで
加え,a) の操作を行ってそれぞれの濃度の標準液について得た指示値の比を補正し,測定対象元素
の指示値と内標準元素の指示値との比の関係線を作成する。
24.8.5
留意事項
a) 各元素の質量数を設定するには,表14を参考するとよい。
b) 妨害物質の存在が不明の場合には,21.6.5 d) による。
c) 安定同位体がある場合,21.6.5 e) による。
d) 定量下限付近の定量で,水による指示値が無視できない場合には,21.6.5 f) による。
25
亜鉛(Zn)
25.1
一般事項
亜鉛の定量には,フレーム原子吸光法,電気加熱原子吸光法,ICP発光分光分析法又はICP質量分析法
を適用する。
25.2
フレーム原子吸光法
試料を前処理した後,アセチレン-空気フレーム中に噴霧し,亜鉛による原子吸光を波長213.9 nmで測
定して,亜鉛を定量する。
定量範囲:Zn:0.05〜2 mg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
25.2.1
試薬
試薬は,次による。
a) 水 24.2.1 a) による。
b) 硝酸(1+1) 21.6.1 b) による。
c) 亜鉛標準液(Zn:10 mg/L) 24.7.1 d) の亜鉛標準液(Zn:100 mg/L)10 mLを全量フラスコ100 mL
にとり,硝酸(1+1)2 mLを加えた後,水を標線まで加える。
25.2.2
器具及び装置
器具及び装置は,次による。
a) フレーム原子吸光分析装置 JIS K 0121で規定するフレーム原子吸光分析装置で,測定対象元素用の
光源を備え,かつ,バックグラウンド補正が可能なもの。
25.2.3
準備操作
準備操作は,次による。
a) 試料を6.6によって前処理する。
b) 空試験としてa) の準備操作での試料と同量の水をとり,試料と同様にa) の操作を行う。
c) 試料中のナトリウム,カリウム,カルシウム,マグネシウムなどの濃度が高く,亜鉛の濃度が低い試
料で,抽出操作を妨害する物質を含まない場合の準備操作は,24.5.3による。
25.2.4
操作
操作は,次による。
a) 25.2.3 a) の準備操作を行った試料を,JIS K 0121の8.(操作方法)の操作に準じてフレーム中に噴霧
し,波長213.9 nmの指示値(吸光度又はその指示値)を読み取る。
b) 空試験として25.2.3 b) の準備操作の水を試料と同様にa) の操作を行って試料について得た指示値を
126
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
補正する。
c) あらかじめ,次によって作成した検量線から亜鉛の濃度を求め,試料中の亜鉛銅の濃度(mg/L)を算
出する。
1) 亜鉛標準液(Zn:10 mg/L)0.5〜20 mLを全量フラスコ100 mLに段階的にとり,25.2.3 a) を行った
試料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。これらの溶液についてa) の
操作を行う。
2) 別に,空試験としてこの操作に用いた水について25.2.3 a) を行った試料と同じ酸の濃度になるよう
に酸を加えた後,a) の操作を行ってそれぞれの標準液について得た指示値を補正し,亜鉛(Zn)の
濃度と指示値との関係線を作成する。検量線の作成は,試料測定時に行う。
3) 準備操作として溶媒抽出を適用するときは,亜鉛標準液(Zn:10 mg/L)の量を,適宜,減らす。24.5.3
のc) 及びd) の抽出によって準備操作を行い,酢酸ブチル層,4-メチル-2-ペンタノン層をそのまま
噴霧する場合の検量線は,亜鉛標準液(Zn:10 mg/L)を適切な濃度(Zn:0.1〜1 mg/L)に薄め,
その2〜40 mLを段階的にとり,500 mL(又は100〜500 mLの一定量)とした後,試料と同様にa) 及
びb) を行って亜鉛(Zn)の濃度と指示値との関係線を作成する。
25.2.5
留意事項
24.5.5による。
25.3
電気加熱原子吸光法
試料を前処理した後,電気加熱炉で原子化し,亜鉛による原子吸光を波長213.9 nmで測定して亜鉛を定
量する。
定量範囲:Zn:1〜20 µg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
この方法は,共存する酸,塩の種類及び濃度の影響を受けやすいので,これらの影響の少ない試料(例
えば,貫流ボイラの給水,揮発性物質処理を行っている水管ボイラの給水及びボイラ水の試料。)に適用す
る。
25.3.1
試薬
試薬は,次による。
a) 水 24.4.1 a) による。
b) 硝酸(1+1) 21.6.1 b) による。
c) 亜鉛標準液(Zn:1 mg/L) 25.2.1 c) の亜鉛標準液(Zn:10 mg/L)10 mLを全量フラスコ100 mLに
とり,硝酸(1+1)2 mLを加えた後,水を標線まで加える。
25.3.2
器具及び装置
器具及び装置は,次による。
a) 電気加熱原子吸光分析装置 JIS K 0121で規定する電気加熱原子吸光分析装置で,測定対象元素用の
光源を備え,かつ,バックグランド補正が可能なもの。
b) マイクロピペット JIS K 0970で規定するピストン式ピペット10〜50 µL,又は自動注入装置。
25.3.3
準備操作
準備操作は,次による。
a) 試料を6.6によって前処理する。
b) 空試験としてa) の準備操作での試料と同量の水をとり,試料と同様にa) の操作を行う。
25.3.4
操作
操作は,次による。
127
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) 25.3.3 a) の準備操作を行った試料の一定量(例えば,10〜50 µL)をマイクロピペットで発熱体に注
入し,JIS K 0121の8.(操作方法)の操作に準じて,乾燥(100〜120 ℃,30〜40秒間)した後,灰
化(600〜1 000 ℃,30〜40秒間)し,次に,原子化(2 200〜2 700 ℃,3〜6秒間)し,波長213.9 nm
の指示値(吸光度又はその指示値)を読み取る。この操作を3回繰り返し,指示値が合うことを確認
する。
なお,乾燥,灰化,原子化の温度と時間とは用いる装置,試料の注入量及び共存する塩類の濃度に
よって異なるので,装置及び試料によってこれらの条件の最適化をはかる。
b) 空試験として25.3.3 b) の準備操作の水を試料と同様にa) の操作を行って試料について得た指示値を
補正する。
c) あらかじめ,次によって作成した検量線から亜鉛の濃度を求め,試料中の亜鉛の濃度(µg/L)を算出
する。
1) 亜鉛標準液(Zn:1 mg/L)0.1〜2 mLを全量フラスコ100 mLに段階的にとり,25.3.3 a) を行った試
料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。この溶液についてa) の操作
を行う。
2) 別に,空試験として水について25.3.3 a) を行った試料と同じ酸の濃度となるように酸を加えた後,
a) の操作を行って標準液について得た指示値を補正し,亜鉛(Zn)の濃度と指示値との関係線を作
成する。検量線の作成は,試料測定時に行う。
25.3.5
留意事項
a) 試料中の亜鉛濃度が極低濃度の場合には,試料の一定量(10〜50 μL)を発熱体に注入し,乾燥する。
この操作を繰り返した後,灰化,原子化の操作を行って指示値を読み取る。この場合は,25.3.4 b) の
操作でも同様な回数を注入し,乾燥する。
25.4
ICP発光分光分析法
24.7による。
25.5
ICP質量分析法
24.8による。
26
鉄(Fe)
26.1
一般事項
鉄の定量には,1,10-フェナントロリン吸光光度法,2,4,6-トリ-2-ピリジル-1,3,5-トリアジン(TPTZ)吸
光光度法,フレーム原子吸光法,電気加熱原子吸光法,ICP発光分光分析法又はICP質量分析法を適用す
る。
26.2
1,10-フェナントロリン吸光光度法
微酸性溶液中で塩化ヒドロキシルアンモニウムと1,10-フェナントロリンを加えた後,酢酸アンモニウム
を加えてpHを4〜5に調節し,生成する赤だいだいの鉄(II)錯体の吸光度を測定して鉄を定量する。
定量範囲:吸収セル10 mmの場合Fe:0.2〜5 mg/L,繰返し精度:2〜10 %
26.2.1
試薬
試薬は,次による。
a) 水 24.4.1 a) による。
b) 塩酸(1+1) 21.5.1 b) による。
c) 硝酸(1+1) JIS K 8541で規定する硝酸を用いて調製する。
128
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
d) アンモニア水(1+1) 24.2.1 b) による。
e) 塩化ヒドロキシルアンモニウム溶液(100 g/L) 22.2.1 d) による。
f)
1,10-フェナントロリン溶液(1 g/L) JIS K 8202で規定する塩化1,10-フェナントロリニウム一水和
物1.3 gを水に溶かして1 Lとする。または,JIS K 8789で規定する1,10-フェナントロリン一水和物
1.1 gをJIS K 8102で規定するエタノール(95)100 mLに溶かし,水を加えて1 Lとする。
g) 酢酸アンモニウム溶液(500 g/L) JIS K 8359で規定する酢酸アンモニウム500 gを水に溶かして1 L
とする。
h) 鉄標準液(Fe:1 000 mg/L) 24.7.1 f) による。
i)
鉄標準液(Fe:10 mg/L) 鉄標準液(Fe:1 000 mg/L)10 mLを全量フラスコ1 000 mLにとり,塩
酸(1+1)20 mLを加えた後,水を標線まで加える。
26.2.2
器具及び装置
器具及び装置は,次による。
a) 光度計 分光光度計又は光電光度計。
26.2.3
準備操作
準備操作は,次による。
a) 試料100 mLについて箇条6の操作を行い,液量を約60 mL以下にする。
b) 試料の鉄濃度が5 mg/L以上の場合は,試料量を減らして操作を行う。
26.2.4
操作
操作は,次による。
a) 26.2.3で前処理した試料の全量をビーカにとる。ただし,試料に有機物及び濁りが少なく,また,妨
害物質が共存しない場合には,d) 以下の操作を行って定量してもよい。
b) 水を加えて50〜100 mLとした後,アンモニア水(1+1)を加えて微アルカリ性とする。これを数分
間煮沸して沈殿を生成させ,しばらく静置する。鉄の量が極めて微量の場合(Fe:20 µg以下)には,
捕集剤としてJIS K 8255で規定する硫酸カリウムアルミニウム・12水0.1 gを加えて溶かし,再びア
ンモニア水(1+1)を加えて微アルカリ性とし,水酸化アルミニウムを生成させ,鉄を完全に捕集し
てろ別する。この際,沈殿は塩酸に溶けにくくなるので,塩酸の添加量を多くし,液量が約5 mLに
なるまで加熱濃縮する。次に,水で液量を約70 mLとし,JIS K 8536で規定する(+)-酒石酸ナトリウ
ムカリウム四水和物0.1 gを加える。以下,d) 以下の操作を行う。
c) 沈殿が沈降した後,ろ紙5種Aでろ過し,温水で数回洗う。沈殿は元のビーカに洗い入れ,塩酸(1
+1)4 mLを加え,加熱して溶かし,先のろ紙でろ過し,同時にろ紙に付着している水酸化鉄(III)
を溶かす。ろ紙は温水で数回洗う。
d) ろ液と洗液とを合わせ,水を加えて液量を約70 mLとした後,塩化ヒドロキシルアンモニウム溶液(100
g/L)1 mLを加えて振り混ぜる。JIS K 9502で規定するL(+)-アスコルビン酸0.1 gを加えてもよい。
e) 1,10-フェナントロリン溶液(1 g/L)5 mLを加えて振り混ぜ,酢酸アンモニウム溶液(500 g/L)10 mL
を加えて振り混ぜ,放冷する。発色時のpHは約4.8になる。塩酸の濃度が高い場合には,アンモニア
水(1+1)による中和操作を行い,発色時のpHを4〜5に調節する。また,pH調節の操作は操作順
序に従い,1,10-フェナントロリン溶液(1 g/L)を加えた後に行う。
f)
全量フラスコ100 mLに移し入れ,水を標線まで加えて約20分間放置する。
g) この溶液の一部を吸収セルに移し,波長510 nm付近の吸光度を測定する。
h) 空試験として試料と同量の水をとり,試料に適用した箇条6の操作を行って空試験の溶液とし,a) と
129
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
同量をビーカにとり硝酸(1+1)1〜2 mLを加えた後,b)〜g) の操作を行って吸光度を測定し,試料
について得た吸光度を補正する。
i)
あらかじめ,次によって作成した検量線から鉄の濃度を求め,試料中の鉄の濃度(mg/L)を算出する。
1) 鉄標準液(Fe:10 mg/L)2〜50 mLを全量フラスコ100 mLに段階的にとり,塩酸(1+1)4 mLを
加え,d)〜h) の操作を行って鉄(Fe)の濃度と吸光度との関係線を作成する。
26.2.5
留意事項
a) 鉄をあらかじめ水酸化物として分離しないで試験する場合には,水銀,銅,カドミウム,ニッケル,
コバルト,亜鉛などが妨害する。ただし,カドミウムは50 mg/L,亜鉛は10 mg/Lまでは妨害しない。
b) pH 3.5で発色させれば,銅は10 mg/L,コバルトは10 mg/Lまでは妨害しない。
c) ニッケルが10 mg/L程度共存する場合は,EDTA溶液(JIS K 8107で規定するエチレンジアミン四酢
酸二水素二ナトリウム二水和物3.7 gを水100 mLに溶かす。)5 mLを添加し,約10分間煮沸すれば
妨害しない。
d) 多量の亜鉛が共存するときは,pH 9で1,10-フェナントロリン溶液(1 g/L)を多量に加えて発色させ
れば,妨害を除くことができる。りん酸イオンが多量に共存すると妨害するが,発色時のpHを5〜7
に調節し,約2時間放置した後,吸光度を測定すれば妨害は少なくなる。
e) 鉄(III)イオンを鉄(II)イオンに還元する場合,溶液の酸性度は0.2〜0.3 mol/L程度がよい。強酸性
では塩化ヒドロキシルアンモニウムだけが分解されて鉄が完全に還元されない。また,還元剤には塩
化ヒドロキシルアンモニウムのほかにJIS K 9502で規定するL(+)-アスコルビン酸を用いてもよい。
f)
発色操作での試薬の添加の順序は,弱酸性とする酸性度の調節,塩化ヒドロキシルアンモニウム溶液,
1,10-フェナントロリン溶液,緩衝溶液の順とする。この順序を変えると還元反応が遅くなり,完全に
発色するのに長時間を要するか,又は完全には発色しなくなる。
g) 発色はpH 2〜9の範囲で生じるが,一般に,pH 3〜5の範囲において短時間で完結する。pH 7以上で
は長時間を必要とする。最高発色に達する時間はpH 3〜5の範囲で液温20 ℃以上で20〜30分間を要
する。
26.3
2,4,6-トリ-2-ピリジル-1,3,5-トリアジン吸光光度法
微酸性溶液中で塩化ヒドロキシルアンモニウムと2,4,6-トリ-2-ピリジル-1,3,5-トリアジンとを加えた後,
酢酸アンモニウムを加えてpHを4〜5に調節し,生成する青い色の鉄(II)錯体の吸光度を測定して鉄を
定量する。
定量範囲:吸収セル20 mmの場合Fe:0.01〜0.3 mg/L,吸収セル50 mmの場合Fe:0.005〜0.2 mg/L,26.3.3
d) 以下のフィルタ濃縮操作で吸収セル50 mmの場合Fe:0.001〜0.1 mg/L,繰返し精度:2〜10 %
26.3.1
試薬
試薬は,次による。
a) 水 24.4.1 a) による。
b) 塩酸(1+1) JIS K 9902で規定する高純度試薬−塩酸を用いて調製する。
c) 塩酸(3 mol/L) JIS K 9902で規定する高純度試薬−塩酸を用いて調製する。
d) 塩化ヒドロキシルアンモニウム溶液(100 g/L) 22.2.1 d) の塩化ヒドロキシルアンモニウム溶液(100
g/L)をe) の1) 及び2) の操作によって鉄を除いたもの。
e) 酢酸アンモニウム溶液(500 g/L) 26.2.1 g) の酢酸アンモニウム溶液(500 g/L)を,次の操作によっ
て鉄を除いたもの。塩化ヒドロキシルアンモニウム溶液(100 g/L)及び酢酸アンモニウム溶液(500 g/L)
の鉄の除去は,次による。
130
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1) 試薬溶液500 mLにJIS K 8227で規定する過塩素酸ナトリウム一水和物0.25 gと2,4,6-トリ-2-ピリ
ジル-1,3,5-トリアジン溶液(1 mmol/L)5 mL[酢酸アンモニウム溶液の場合には,塩化ヒドロキシ
ルアンモニウム溶液(100 g/L)5 mLを加える。]との割合で加えて振り混ぜた後,分液漏斗に移し
入れ,JIS K 8723で規定するニトロベンゼン25 mLを加えて約2分間激しく振り混ぜて放置する。
2) 着色したニトロベンゼン層を捨てる。2,4,6-トリ-2-ピリジル-1,3,5-トリアジン溶液(1 mmol/L)5 mL
とニトロベンゼン25 mLとを加えて抽出を行う。この操作は,ニトロベンゼン層が着色しなくなる
まで繰り返し行う。
f)
酢酸アンモニウム溶液(0.1 mol/L) e) の酢酸アンモニウム溶液(500 g/L)を用いて調製する。
g) 2,4,6-トリ-2-ピリジル-1,3,5-トリアジン溶液(1 mmol/L) 2,4,6-トリ-2-ピリジル-1,3,5-トリアジン0.16
gを塩酸(1+1)1 mLに溶かし,全量フラスコ500 mLに移し入れ,水を標線まで加える。
h) 鉄標準液(Fe:100 mg/L) 24.7.1 g) による。
i)
鉄標準液(Fe:1 mg/L) h) の鉄標準液(Fe:100 mg/L)5 mLを全量フラスコ500 mLにとり,塩酸
(1+1)10 mLを加えた後,水を標線まで加える。
26.3.2
器具及び装置
器具及び装置は,次による。
a) 光度計 分光光度計又は光電光度計。
b) メンブレンフィルタ PTFE製,直径47 mm,孔径0.45 μm。
c) キレートフィルタ 基材:PTFE,樹脂:イミノ二酢酸(IDA)型,直径47 mm,樹脂サイズ10 μm。
又は,同等の性能をもつもの。
d) 減圧ろ過器 11.3.1 b) のろ過器(分離形)の図6に示す一例と同じであるが,フィルタ通過後の塩酸
を回収するため,吸引瓶はビーカを設置できる開放型が望ましい。
26.3.3
準備操作
準備操作は,次による。
a) 試料100 mLについて箇条6の操作を行い,液量を約20 mL以下にする。
b) 試料の鉄濃度が0.3 mg/L以上の場合は,試料量を減らして操作を行う。
c) 試料の鉄濃度が5 μg/L以下の場合は,d) 以下の操作(フィルタ濃縮法)によって濃縮をする。
d) メンブレンフィルタ及びキレートフィルタを塩酸(3 mol/L)で洗浄し,減圧ろ過用焼結ガラス製フィ
ルタホルダに下からキレートフィルタ,メンブレンフィルタの順に取り付ける。続いて,塩酸(3 mol/L)
を20 mL,水を50 mL,酢酸アンモニウム溶液(0.1 mol/L)50 mLを順に10 mLずつフィルタへ通液
する。この操作は,フィルタに付着している鉄の除去及びキレートフィルタの活性化を目的としてい
る。
e) メスシリンダで試料1 Lをはかりとり,フィルタへ通水して試料中の鉄をフィルタに捕集した後,水
20 mLを通水しフィルタを洗浄する。ただし,試料のpHは5.5以上とし,キレートフィルタで鉄を捕
捉できることを確認しておく。メンブレンフィルタをろ過器からコニカルビーカ(300 mL容量)へ移
し,メンブレンフィルタを入れたコニカルビーカを受器として,キレートフィルタに塩酸(3 mol/L)
を10 mLずつ合計100 mL添加してキレートフィルタで捕集した鉄を溶出させる。このとき,キレー
トフィルタ全面に塩酸が浸せきするようにして溶出させる。
f)
塩酸溶出後,コニカルビーカに時計皿をかぶせ,20 mLまで加熱濃縮する。冷却後,メンブレンフィ
ルタを取り出して水で洗浄し,再度10 mLまで加熱濃縮する。
g) 濃縮後,室温まで放冷させた試料に対し,26.3.4の操作によって鉄濃度を定量する。
131
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
26.3.4
操作
操作は,次による。
a) 26.3.3のa)〜f) で前処理した試料の全量をビーカにとる。
b) 塩化ヒドロキシルアンモニウム溶液(100 g/L)1 mLを加えて振り混ぜ,2,4,6-トリ-2-ピリジル-1,3,5-
トリアジン溶液(1 mmol/L)5 mLを加えて再び振り混ぜる。
c) 酢酸アンモニウム溶液(500 g/L)を加えてpHを4〜5に調節し,全量フラスコ50 mLに水とともに移
し入れ,水を標線まで加え,約5分間放置する。例えば,試料100 mLをとり,箇条6の操作で塩酸5
mLを用いた場合には,酢酸アンモニウム溶液(500 g/L)の量は約15 mL必要である。pH試験紙は,
ブロモクレゾールグリーンを用いる。
d) この溶液の一部を吸収セルに移し,波長595 nm付近の吸光度を測定する。
e) 空試験として試料と同量の水をとり,試料に適用した箇条6の操作を行って空試験用の溶液とし,a)
と同量をビーカにとり,b)〜d) の操作を行って試料について得た吸光度を補正する。
f)
あらかじめ,次によって作成した検量線から鉄の濃度を求め,試料中の鉄の濃度(mg/L)を算出する。
1) 鉄標準液(Fe:1 mg/L)1〜30 mLをビーカに段階的にとり,塩酸(1+1)4 mLを加えて水で液量
を約25 mLとし,b)〜d) の操作を行う。
2) 別に,空試験としてこの操作に用いた水について塩酸(1+4)4 mLを加え,b)〜d) の操作を行い,
標準液について得た吸光度を補正し,鉄(Fe)の濃度と吸光度との関係線を作成する。
26.4
フレーム原子吸光法
試料を前処理した後,アセチレン-空気フレーム中に噴霧し,鉄による原子吸光を波長248.3 nmで測定
して鉄を定量する。
定量範囲:Fe:0.3〜6 mg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
26.4.1
試薬
試薬は,次による。
a) 水 24.4.1 a) による。
b) 鉄標準液(Fe:10 mg/L) 26.2.1 i) による。
26.4.2
器具及び装置
器具及び装置は,次による。
a) フレーム原子吸光分析装置 JIS K 0121で規定するフレーム原子吸光分析装置で,測定対象元素用の
光源を備え,かつ,バックグラウンド補正が可能なもの。
26.4.3
準備操作
準備操作は,次による。
a) 試料を6.6によって前処理する。
b) 空試験としてa) の準備操作での試料と同量の水をとり,試料と同様にa) の操作を行う。
26.4.4
操作
操作は,次による。
a) 26.4.3 a) の準備操作を行った試料を,JIS K 0121の8.(操作方法)の操作に準じてフレーム中に噴霧
し,波長248.3nmの指示値(吸光度又はその指示値)を読み取る。
b) 空試験として26.4.3 b) の準備操作の水を試料と同様にa) の操作を行って試料について得た指示値を
補正する。
c) あらかじめ,次によって作成した検量線から鉄の濃度を求め,試料中の鉄の濃度(mg/L)を算出する。
132
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1) 鉄標準液(Fe:10 mg/L)3〜60 mLを全量フラスコ100 mLに段階的にとり,26.4.3 a) を行った試料
と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。この溶液についてa) の操作を
行う。
2) 別に,空試験として水について26.4.3 a) を行った試料と同じ酸の濃度になるように酸を加えた後,
a) の操作を行って標準液について得た指示値を補正し,鉄(Fe)の濃度と指示値との関係線を作成
する。検量線の作成は,試料測定時に行う。
26.4.5
留意事項
a) シリカを多量に含む試料の場合には,干渉抑制剤としてカルシウム(又はマグネシウム)を200 mg/L
程度加えておく。
26.5
電気加熱原子吸光法
試料を前処理した後,電気加熱炉で原子化し,鉄による原子吸光を波長248.3 nmで測定して鉄を定量す
る。
定量範囲:Fe:5〜100 µg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
この方法は,共存する酸,塩の種類及び濃度の影響を受けやすいので,これらの影響の少ない試料(例
えば,貫流ボイラの給水,揮発性物質処理を行っている水管ボイラの給水及びボイラ水の試料)に適用す
る。
26.5.1
試薬
試薬は,次による。
a) 水 24.4.1 a) による。
b) 硝酸(1+1) 21.6.1 b) による。
c) 鉄標準液(Fe:1 mg/L) 26.2.1 i) の鉄標準液(Fe:10 mg/L)10 mLを全量フラスコ100 mLにとり,
硝酸(1+1)2 mLを加え,水を標線まで加える。
26.5.2
器具及び装置
器具及び装置は,次による。
a) 電気加熱原子吸光分析装置 JIS K 0121で規定する電気加熱原子吸光分析装置で,測定対象元素用の
光源を備え,かつ,バックグランド補正が可能なもの。
b) マイクロピペット JIS K 0970で規定するピストン式ピペット10〜50 µL,又は自動注入装置
26.5.3
準備操作
準備操作は,次による。
a) 試料を6.6によって前処理する。
b) 空試験としてa) の準備操作での試料と同量の水をとり,試料と同様にa) の操作を行う。
26.5.4
操作
操作は,次による。
a) 26.5.3 a) の準備操作を行った試料の一定量(例えば,10〜50 µL)をマイクロピペットで発熱体に注
入し,JIS K 0121の8.(操作方法)の操作に準じて,乾燥(100〜120 ℃,30〜40秒間)した後,灰
化(600〜1 000 ℃,30〜40秒間)し,次に,原子化(2 200〜2 800 ℃,3〜6秒間)し,波長248.3 nm
の指示値(吸光度又はその指示値)を読み取る。この操作を3回繰り返し,指示値が合うことを確認
する。
なお,乾燥,灰化,原子化の温度と時間は用いる装置,試料の注入量及び共存する塩類の濃度によ
って異なるので,装置や試料によってこれらの条件の最適化をはかる。

133
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) 空試験として26.5.3 b) の準備操作の水を試料と同様にa) の操作を行って試料について得た指示値を
補正する。
c) あらかじめ,次によって作成した検量線から鉄の濃度を求め,試料中の鉄の濃度(µg/L)を算出する。
1) 鉄標準液(Fe:1 mg/L)0.5〜10 mLを全量フラスコ100 mLに段階的にとり,26.5.3 a) を行った試
料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。これらの溶液についてa) の
操作を行う。
2) 別に,空試験としてこの操作に用いた水について26.5.3 a) を行った試料と同じ酸の濃度になるよう
に酸を加えた後,a) の操作を行ってそれぞれの標準液について得た指示値を補正し,鉄(Fe)の濃
度と指示値との関係線を作成する。検量線の作成は,試料測定時に行う。
26.5.5
留意事項
a) 試料中の鉄濃度が極低濃度の場合には,試料の一定量(10〜50 μL)を発熱体に注入し,乾燥する。こ
の操作を繰り返した後,灰化,原子化の操作を行って指示値を読み取る。この場合は,26.5.4 b) の操
作でも同様な回数を注入し,乾燥する。
26.6
ICP発光分光分析法
24.7による。
26.7
ICP質量分析法
試料を前処理した後,内標準元素(イットリウム,インジウム,ビスマス)を加え,試料導入部を通し
て高周波プラズマ中に噴霧し,鉄及び内標準元素のそれぞれの質量/電荷数における指示値(イオンカウン
ト値又はその比例値)を測定し,鉄の指示値と内標準元素の指示値との比を求めて鉄を定量する。
表15−定量範囲,繰返し精度及び質量数の例a)
対象元素
濃度
繰返し精度
質量数
μg/L
%
鉄(Fe)
0.5〜500
2〜10
56,54,57
イットリウム(Y)b)
−
−
89
インジウム(In)b)
−
−
115
ビスマス(Bi)b)
−
−
209
注a) 装置及び測定条件によって異なる。
b) 内標準元素
26.7.1
試薬
試薬は,次による。
a) 水 24.4.1 a) による。
b) 硝酸(1+1) 21.6.1 b) による。
c) 鉄標準液(Fe:1 mg/L) 26.5.1 c) による。
d) 内標準液(1 mg/L) 内標準元素としてイットリウム,インジウム又はビスマスを用いる。内標準液
の調製には,21.6.1のe)〜g) で規定する溶液のうち内標準とする元素の溶液2 mLを全量フラスコ100
mLにとり,硝酸(1+1)2 mLを加え,水を標線まで加える。使用時に調製する。
26.7.2
器具及び装置
器具及び装置は,次による。
a) ICP質量分析計 JIS K 0133による。ICP質量分析計は,カルシウム,鉄などの定量において,スペ
134
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
クトル干渉となるAr(40),ArO(56),CaO(56)などの分子イオンのピークによる干渉を低減でき
るものを用いる。干渉を低減する方法としてプラズマの温度を低くする方式の装置がある。また,コ
リジョン・リアクションセルなどのスペクトル干渉除去機能が搭載されている装置もある。
26.7.3
準備操作
準備操作は,次による。
a) 試料を6.6によって前処理し,全量フラスコ100 mLに移し入れ,いずれかの内標準液(1 mg/L)1 mL
を加え,硝酸の最終濃度が0.1〜0.5 mol/Lになるように硝酸(1+1)を加えた後,水を標線まで加え
る。このときの濃度が表15の定量範囲になるようにする。
b) 空試験の溶液として同量の水を6.6によって前処理し,全量フラスコ100 mLに移し入れ,a) と同じ
内標準溶液(1 mg/L)1 mLを加え,試料と同じ酸の濃度になるように硝酸(1+1)を加えた後,水を
標線まで加える。
c) JIS K 0133の箇条8(分析装置の最適化)及び箇条9(分析条件の決定)に準じて,ICP質量分析計を
定量できる状態に調整する。
26.7.4
操作
操作は,次による。
a) 26.7.3 a) の試料をJIS K 0133の箇条12(定量分析)に準じて,試料導入部を通して高周波プラズマ
中に噴霧して測定対象元素と内標準物質との質量/電荷数における指示値(イオン電流又はその比例
値)を読み取り,測定対象元素の指示値と内標準物質の指示値との比を求める。
b) 空試験として26.7.3 b) の溶液についてa) の操作を行って測定対象元素の指示値と内標準物質の指示
値との比を求め,試料について得た測定対象元素と内標準物質との指示値の比を補正する。
c) 検量線から測定対象元素の濃度を求め,試料中の測定対象元素の濃度(ng/L)(又はµg/L)を算出す
る。
d) 銅と亜鉛を同時定量する場合には,それぞれの金属元素について,それぞれの検量線を作成する。検
量線の作成は,試料測定時に行う。
1) 混合標準液[(Fe:1 mg,Cu:1 mg,Zn:1 mg)/L]0.05〜50 mLを全量フラスコ100 mLに段階的に
とり,26.7.3 a) と同じ内標準溶液(1 mg/L)1 mLを加え,26.7.3 a) の試料と同じ酸の濃度になるよ
うに硝酸(1+1)を加え,水を標線まで加える。
2) 別に,空試験としてこの操作に用いた水を全量フラスコ100 mLにとり,内標準溶液(1 mg/L)1 mL
を加え,26.7.3 a) の試料と同じ酸の濃度になるように硝酸(1+1)を加えた後,同じ水を標線まで
加え,a) の操作を行ってそれぞれの標準液について得た指示値の比を補正し,測定対象元素の指示
値と内標準元素の指示値との比の関係線を作成する。
26.7.5
留意事項
a) 銅及び亜鉛の質量数を設定するには,表14を参考するとよい。
b) 妨害物質の存在が不明の場合には,21.6.5 d) による。
c) 安定同位体がある場合,21.6.5 e) による。
d) 定量下限付近の定量で,水による指示値が無視できない場合には,21.6.5 f) による。

135
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
付表1 引用規格
JIS B 7411-1
JIS B 7411-2
JIS B 8223
JIS C 1602
JIS C 1604
JIS C 1611
JIS G 3454
JIS G 3456
JIS G 3459
JIS G 3463
JIS H 6201
JIS H 6202
JIS K 0050
JIS K 0094
JIS K 0101
JIS K 0102
JIS K 0115
JIS K 0116
JIS K 0121
JIS K 0122
JIS K 0126
JIS K 0127
JIS K 0133
JIS K 0211
JIS K 0215
JIS K 0552
JIS K 0557
JIS K 0802
JIS K 0805
JIS K 0970
JIS K 1101
JIS K 1107
JIS K 8005
JIS K 8019
JIS K 8034
JIS K 8051
JIS K 8061
JIS K 8085
一般用ガラス製温度計−第1部:一般計量器
一般用ガラス製温度計−第2部:取引又は証明用
ボイラの給水及びボイラ水の水質
熱電対
測温抵抗体
サーミスタ測温体
圧力配管用炭素鋼鋼管
高温配管用炭素鋼鋼管
配管用ステンレス鋼鋼管
ボイラ・熱交換器用ステンレス鋼鋼管
化学分析用白金るつぼ
化学分析用白金皿
化学分析方法通則
工業用水・工場排水の試料採取方法
工業用水試験方法
工場排水試験方法
吸光光度分析通則
発光分光分析通則
原子吸光分析通則
イオン電極測定方法通則
流れ分析通則
イオンクロマトグラフィー通則
高周波プラズマ質量分析通則
分析化学用語(基礎部門)
分析化学用語(分析機器部門)
超純水の電気伝導率試験方法
用水・排水の試験に用いる水
pH自動計測器
有機体炭素(TOC)自動計測器
ピストン式ピペット
酸素
窒素
容量分析用標準物質
亜硝酸ナトリウム(試薬)
アセトン(試薬)
3-メチル-1-ブタノール(試薬)
亜硫酸ナトリウム(試薬)
アンモニア水(試薬)

136
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
付表1 引用規格(続き)
JIS K 8092
JIS K 8101
JIS K 8102
JIS K 8103
JIS K 8107
JIS K 8116
JIS K 8121
JIS K 8129
JIS K 8142
JIS K 8150
JIS K 8155
JIS K 8180
JIS K 8201
JIS K 8202
JIS K 8223
JIS K 8227
JIS K 8228
JIS K 8230
JIS K 8253
JIS K 8255
JIS K 8271
JIS K 8284
JIS K 8295
JIS K 8322
JIS K 8355
JIS K 8359
JIS K 8371
JIS K 8377
JIS K 8432
JIS K 8443
JIS K 8454
JIS K 8474
JIS K 8496
JIS K 8506
JIS K 8519
JIS K 8522
JIS K 8532
JIS K 8533
インジゴカルミン(試薬)
エタノール(99.5)(試薬)
エタノール(95)(試薬)
ジエチルエーテル(試薬)
エチレンジアミン四酢酸二水素二ナトリウム二水和物(試薬)
塩化アンモニウム(試薬)
塩化カリウム(試薬)
塩化コバルト(II)六水和物(試薬)
塩化鉄(III)六水和物(試薬)
塩化ナトリウム(試薬)
塩化バリウム二水和物(試薬)
塩酸(試薬)
塩化ヒドロキシルアンモニウム(試薬)
塩化1,10-フェナントロリニウム一水和物(試薬)
過塩素酸(試薬)
過塩素酸ナトリウム一水和物(試薬)
過塩素酸マグネシウム(試薬)
過酸化水素(試薬)
ペルオキソ二硫酸カリウム(試薬)
硫酸カリウムアルミニウム・12水(試薬)
キシレン(試薬)
くえん酸水素二アンモニウム(試薬)
グリセリン(試薬)
クロロホルム(試薬)
酢酸(試薬)
酢酸アンモニウム(試薬)
酢酸ナトリウム三水和物(試薬)
酢酸ブチル(試薬)
酸化マグネシウム(試薬)
シアン化カリウム(試薬)
N,N-ジエチルジチオカルバミド酸ナトリウム三水和物(試薬)
二しゅう酸三水素カリウム二水和物(試薬)
p-ジメチルアミノベンズアルデヒド(試薬)
臭化カリウム(試薬)
しゅう酸二水和物(試薬)
しゅう酸カリウム一水和物(試薬)
L(+)-酒石酸(試薬)
ビス[(+)-タルトラト]二アンチモン(III)酸二カリウム三水和物(試薬)

137
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
付表1 引用規格(続き)
JIS K 8536
JIS K 8541
JIS K 8548
JIS K 8550
JIS K 8574
JIS K 8576
JIS K 8588
JIS K 8603
JIS K 8617
JIS K 8622
JIS K 8625
JIS K 8637
JIS K 8646
JIS K 8659
JIS K 8663
JIS K 8721
JIS K 8723
JIS K 8736
JIS K 8776
JIS K 8780
JIS K 8789
JIS K 8799
JIS K 8809
JIS K 8810
JIS K 8819
JIS K 8824
JIS K 8830
JIS K 8839
JIS K 8840
JIS K 8848
JIS K 8866
JIS K 8889
JIS K 8891
JIS K 8893
JIS K 8896
JIS K 8903
JIS K 8905
JIS K 8913
(+)-酒石酸ナトリウムカリウム四水和物(試薬)
硝酸(試薬)
硝酸カリウム(試薬)
硝酸銀(試薬)
水酸化カリウム(試薬)
水酸化ナトリウム(試薬)
アミド硫酸アンモニウム(試薬)
ソーダ石灰(試薬)
炭酸カルシウム(試薬)
炭酸水素ナトリウム(試薬)
炭酸ナトリウム(試薬)
チオ硫酸ナトリウム五水和物(試薬)
デキストリン水和物(試薬)
でんぷん(溶性)(試薬)
2,2',2''-ニトリロトリエタノール(試薬)
p-ニトロフェノール(試薬)
ニトロベンゼン(試薬)
エリオクロムブラックT(試薬)
2-ヒドロキシ-1-(2-ヒドロキシ-4-スルホ-1-ナフチルアゾ)-3-ナフトエ酸(試薬)
ピロガロール(試薬)
1,10-フェナントロリン一水和物(試薬)
フェノールフタレイン(試薬)
フタル酸水素カリウム(試薬)
1-ブタノール(試薬)
ふっ化水素酸(試薬)
D(+)-グルコース(試薬)
ウラニン(試薬)
2-プロパノール(試薬)
ブロモクレゾールグリーン(試薬)
ヘキサン(試薬)
四ほう酸ナトリウム十水和物(試薬)
メタクレゾールパープル(試薬)
メタノール(試薬)
メチルオレンジ(試薬)
メチルレッド(試薬)
4-メチル-2-ペンタノン(試薬)
七モリブデン酸六アンモニウム四水和物(試薬)
よう化カリウム(試薬)
138
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
付表1 引用規格(続き)
JIS K 8920
JIS K 8951
JIS K 8953
JIS K 8960
JIS K 8962
JIS K 8979
JIS K 8982
JIS K 8983
JIS K 8987
JIS K 9005
JIS K 9007
JIS K 9020
JIS K 9047
JIS K 9062
JIS K 9502
JIS K 9704
JIS K 9901
JIS K 9902
JIS K 9904
JIS K 9905
JIS P 3801
JIS R 1301
JIS R 1302
JIS R 3503
JIS R 3505
JIS Z 0701
JIS Z 1703
JIS Z 8802
よう素(試薬)
硫酸(試薬)
硫酸亜鉛七水和物(試薬)
硫酸アンモニウム(試薬)
硫酸カリウム(試薬)
硫酸アンモニウム鉄(II)六水和物(試薬)
硫酸アンモニウム鉄(III)・12水(試薬)
硫酸銅(II)五水和物(試薬)
硫酸ナトリウム(試薬)
りん酸(試薬)
りん酸二水素カリウム(試薬)
りん酸水素二ナトリウム(試薬)
L-グルタミン酸(試薬)
ニッケル(試薬)
L(+)-アスコルビン酸(試薬)
2-アミノ-2-ヒドロキシメチル-1,3-プロパンジオール(試薬)
高純度試薬−硝酸
高純度試薬−塩酸
高純度試薬−過塩素酸
高純度試薬−硫酸
ろ紙(化学分析用)
化学分析用磁器るつぼ
化学分析用磁器蒸発ざら
化学分析用ガラス器具
ガラス製体積計
包装用シリカゲル乾燥剤
ポリエチレンびん
pH測定方法
139
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書A
(参考)
試料及び試料採取
A.1 試料及び試料採取
この規格では,5.1(試料),5.2(試料採取),5.3(試料の取扱い),5.4(試料の保存処理),及び5.5(試
験時期)の五つに区分した。
A.1.1 試料
この規格では,各種の試験を行うために,試料は,給水及びボイラ水から採取した水を指し,試験目的
に合致したそれぞれの採取位置における給水及びボイラ水を代表できるものとしている。
A.1.2 試料採取
試料採取について共通する事項については,JIS K 0094,及びJIS K 0410-3-7で規定しているが,この
規格では,次のように区分した。
5.2.1
試料容器
5.2.2
試料採取位置
5.2.3
試料採取装置
5.2.4
試料採取操作
5.2.5
試料採取時の記録事項
5.2.6
留意事項
a) 試料容器 試料容器は,無色共栓ほうけい酸ガラス瓶(ほうけい酸ガラス−1)又は共栓ポリエチレン
瓶(又は共栓ポリプロピレン瓶など。)を用いる。無色共栓ほうけい酸ガラス瓶は試料容器として好適
である。試料の観察が容易であり,試料の変質が少ないなど多くの利点がある。しかし,衝撃に弱く,
多量の試料の運搬には不便であり,また,微量ではあるがほうけい酸ガラスの成分としてシリカ,ナ
トリウム,カリウム,ほう素,アルミニウムなどが溶出する。製品によっては,ひ素,アンチモン,
亜鉛なども溶出する。試験目的によってはA3の水又はA2の水を入れて数日間放置して溶出物を試験
する。
共栓ポリエチレン瓶は,試料容器として最も多く用いられている。衝撃に強く,軽便であり,耐薬
品性に優れている。一般に,瓶からの溶出も少ないが,製品によっては製造時に使用するモリブデン,
クロム,チタンなどの重金属元素が溶出する場合もある。共栓ポリエチレン瓶は,重金属元素,りん
酸イオン,有機物などを吸着する傾向があり,試験目的によっては好ましくない。また,洗浄の際,
塩酸,硝酸などで洗浄すると,その後の水洗によっても酸分がなかなか除去されない。その他の薬品
を用いて洗浄した場合も同様であるので,十分に水で洗浄して付着物を除去する。
なお,ポリエチレンは有機溶媒に侵されやすいので注意する。
溶存酸素,亜硫酸イオン,ヘキサン抽出物質の試験に用いる試料容器はそれぞれの試験方法で規定
してある。
b) 試料採取位置 給水,ボイラ水及び蒸気の試料は,特に断らない限り,その系統中の試験成分の濃度
が最も高い値を示す位置,又は試験目的に最も適した位置から採取しなければならない。
採取位置についてはJIS B 8223で規定する位置とした。
1) 給水の試料採取位置 ボイラの種類に応じた試料採取位置の一例を図A.1〜図A.3に示す。このう

140
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ち特殊循環ボイラのものはこのほかにも数種類あり,基本的には大きな違いはないが,試料の採取
に当たってはボイラ系統に照らして試験目的に合致した場所から採取することが望ましい。系統図
の中で丸ボイラを示していないが,系統的には水管ボイラ(循環形)と同一と考えられるので省略
した。
2) ボイラ水の試料採取位置 丸ボイラにはボイラ水の試料採取装置が設置されていないものもある
が,この場合は水面計のドレン管から採取しても実質的には問題ないと考えられる。
3) 蒸気の試料採取位置 蒸気試料を採取するノズルの取付位置は,特に断らない限り,蒸気が蒸気管
の内径の10倍以上の内径変化のない直管部を流れた位置で,かつ,その位置の直後の至近距離に曲
管部及び内径の変化,その他,蒸気の流れを乱すような条件のないところとする。また,蒸気中の
粒子状物質の存在を考慮,指向性ノズルを用いて等速サンプリングすることが望ましい。
図A.1−水管ボイラの試料採取位置の一例
プラント
蒸気出口
S-6
S-5
火炉
S-4
エコノマイザ
C.T.M
薬液注入装置
試料採取箇所
S
S-2
S-1
S-3
ボイラ
給水ポンプ
脱気器
給水
タンク
薬液注入
脱気器
給水ポンプ
低圧給水加熱器
過熱器
薬液注入
141
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図A.2−特殊循環(単管)ボイラの試料採取位置の一例
図A.3−貫流ボイラの試料採取位置の一例
c) 試料採取装置 給水,ボイラ水及び蒸気の試料を採取する場合には,試料の冷却部及び減圧系統は,
一般に図A.4による。ただし,試料が室温,圧力2 MPa以下ならば冷却器及び減圧装置を省略するこ
とができる。
S-1
プラント
蒸気出口
薬液注入
S-3
S-2
火炉
S
薬液注入装置
汽
水
分
離
器
給水
ボイラ
給水ポンプ
脱
気
タ
ン
ク
試料採取箇所
試料採取装置
薬液注入装置
復水検塩装置
純粋装置
C.T.M
C.H.M
C.M
過熱器
再熱器
S.NA
純水装置
補給水
高圧
タービン 低圧タービン
復水器
復水検塩装置
C.P.O.N.T.M
C.P.T.M
C.T.M
P.O.M
給水
ポンプ
脱気器
C.O.N.T.M
C.M
T.M
C.M
C.M.O.T
給水ポンプ
復水ブースタポンプ
エコノマイザ
S.NA
C.H.M
N2H4注入
NH3注入
火炉
M: 化学分析
C: 電気伝導率
P: pH計
O: 溶存酸素分析計
H: 溶存水素分析計
N: ヒドラジン計
NA: ナトリウム分析計
S: シリカ分析計
T: 濁度計
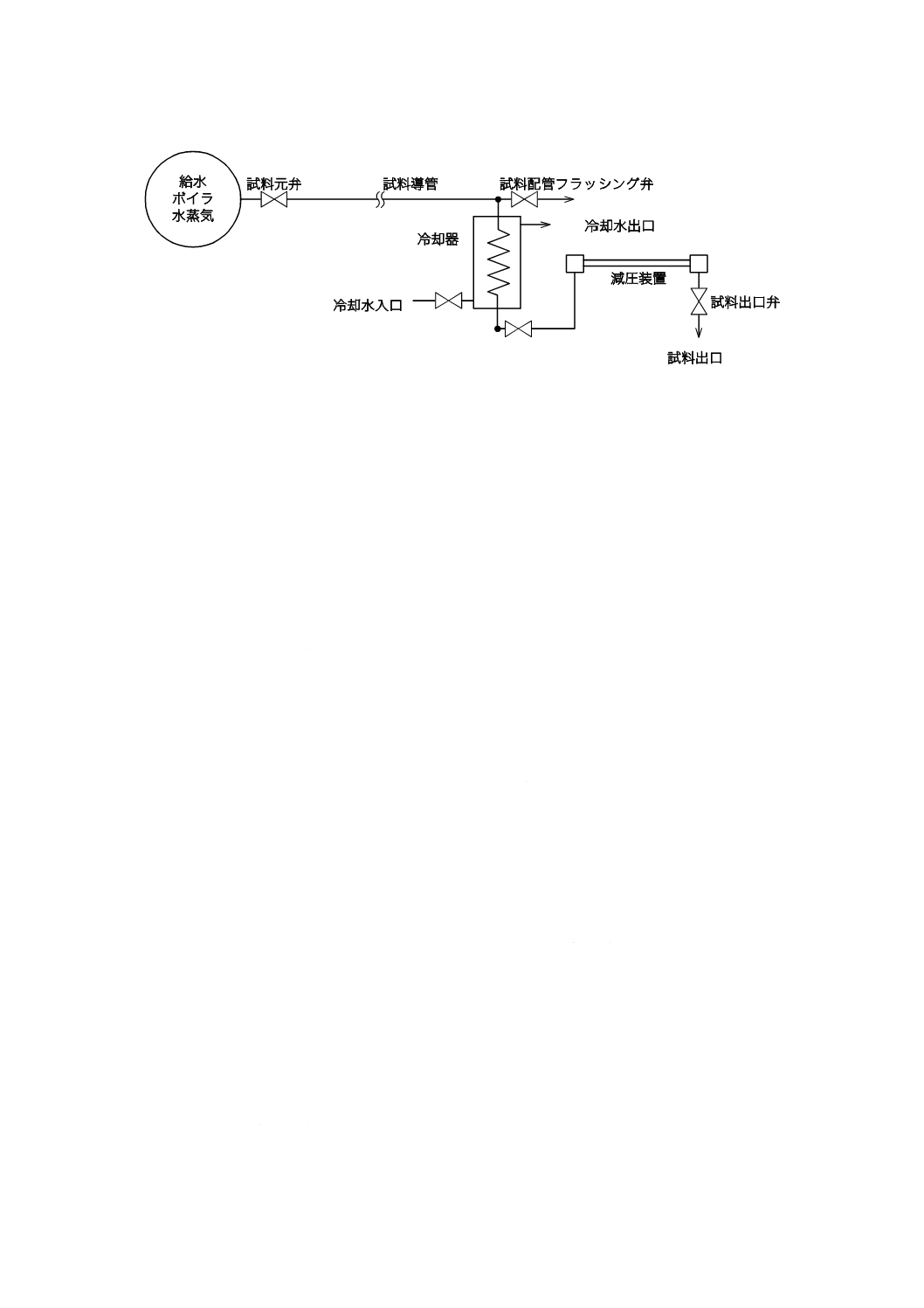
142
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図A.4−試料の冷却部及び減圧系統の一例
給水,ボイラ水及び蒸気の試料を採取する場合には,試料採取位置から試料を試料導管で取り出し,
冷却器及び減圧装置を通して室温より1〜2 ℃低い温度の試料を採取する。試料の冷却及び減圧を行
う場合は,図A.4に示したような方式による。
大形のボイラプラントにおいては,試料採取操作を効率よく行うため冷却器及び減圧装置を適切な
場所に集合させ,それぞれの試料採取位置から採取した試料を試料導管で導いているが,ボイラプラ
ントの規模が大きくなればなるほど試料導管が長くなる。このことは,試料中のイオン状の成分を試
験する場合には,全く問題はないが,試料中の重金属元素及びアルミニウムなどのこれらの一部が懸
濁状で存在している成分を試験する場合には,これらの一部が試料導管中に堆積又は付着することが
ある。したがって,試料導管の長さはできるだけ短くすることが望ましい。さらに,レイノルズ数4 000
を超える乱流条件で試料が流れることを確実にするために,十分に小口径の配管を用いることが望ま
しい。特に,給水中の銅を試験する試料を採取する場合には,注意が必要である。
1) 試料導管 各系統から取り出した試料を冷却装置に導くための管で,材質は,この規格で規定して
いるものを用いる。
圧力が1.96 MPa以上のボイラでは,試料出口弁で調節できる流量の目安は試料出口弁の全開時の
流量に対して次のようである。
ボイラ圧力(MPa)
試料出口弁で調節できる流量範囲(%)
1.96 以下
0〜 100
4.9 以下
0〜 37
7.8 以下
0〜 21
10.8 以下
0〜 15
14.7 以下
0〜 11
17.6 以下
0〜
9
24.5 以下
0〜
6
試料出口弁をこれ以上調節して試料流量を調節したとしても,弁及び弁座が侵食されるおそれが
あるので,圧力が1.96 MPa以上のボイラの場合には,図A.5のような減圧装置を設置しなければな
らない。また,試料導管は,腐食生成物,懸濁物などの堆積及び付着による閉塞を避けるために,
試料元弁から以降の導管の立ち上がり又は水平部分をできるだけ少なくする。特に,溶存酸素を試
験する試料を採取する試料導管には途中に空気が滞留しないように,導管の設置に注意する。
143
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図A.5−固定心線式減圧器の一例
2) 冷却器 二重管コイル式及び浸管コイル式冷却器(図A.6参照)を一般に使用する。冷却の能力は,
室温より約2 ℃低い試料を約1 L/minの流量で採取できるものが望ましい。
試料の冷却に使用する冷却水は,水道水又は清澄な水で,できるだけ温度が低いことが望ましい。
冷却水の温度は室温より5 ℃以上低いものでないと試料の温度を室温以下に冷却することは難し
い。冷却水の温度が高い場合には,一次冷却した水を冷却水として使用するのがよい。
冷却水には腐食性及びスケール生成物のないものを用いるようにすることが望ましい。我が国の
天然水は,そのほとんどが腐食性である。腐食性の強い水を使用する場合には,腐食抑制剤を添加
して腐食を防止するが,腐食抑制剤の過剰添加による障害が生じないように注意する。また,スケ
ールの生成性のある水を用いると,冷却水配管及び冷却器にスケールが生成して冷却効果を阻害し
て多量の冷却水を必要とすることになる。このような場合には,スケール防止剤を用いるが,過剰
添加による障害が生じないように注意する。
冷却器の冷却コイルに付着するスケール又は腐食生成物の程度は,使用する冷却水の水質によっ
て著しく異なるが,一般には,2〜3年間経過したものは冷却コイルの熱伝導率が設置当初と比較し
て20 %程度低下するので,2〜3年間ごとに冷却コイルの化学洗浄を行うとよい。
144
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図A.6−浸管コイル式冷却器の一例
3) 減圧装置 固定心線式減圧器は,試料の圧力が変動しない場合に用いる。二重心線式減圧器は,試
料の圧力が変動する場合に用いる。この減圧器は,手動操作でステムを回転して可変減圧すること
ができる。
自動減圧装置は,圧力に対応して自動的に可変減圧することができる。
圧力1.97 MPa以上の給水,ボイラ水及び蒸気の試料を採取する場合には,減圧して採取する。減
圧装置は,固定心線式減圧器(図A.5参照),手動操作の二重心線式の減圧器(図A.7参照)及び自
動減圧器(図A.8参照)を用いる。ボイラの圧力を変圧して運転する場合には,手動操作でステム
を回転することによって容易に可変減圧することができる二重心線式減圧器及びボイラの圧力に対
応して自動的に可変減圧することができる自動減圧装置が便利である。
1) の試料導管に記載したように試料出口弁で流出量を調節するのは,圧力が高くなればなるほど
流量の調節が困難になり,更に,弁及び弁座が侵食されるので,1個の弁で流量の調節及び減圧を
行うことはできない。また,高圧ボイラの給水の溶存酸素を試験する試料を採取する場合に,減圧
器を用いずに試料出口弁で極端に減圧すると,この過程において水の一部が分解して酸素と水素と
を生じ,試料中の溶存酸素の濃度が過大に定量されるので,必ず減圧器を使用する。
試料入口
試料出口
外筒
冷却水出口
冷却コイル
(材料はSUS)
冷却水入口
(SGP)
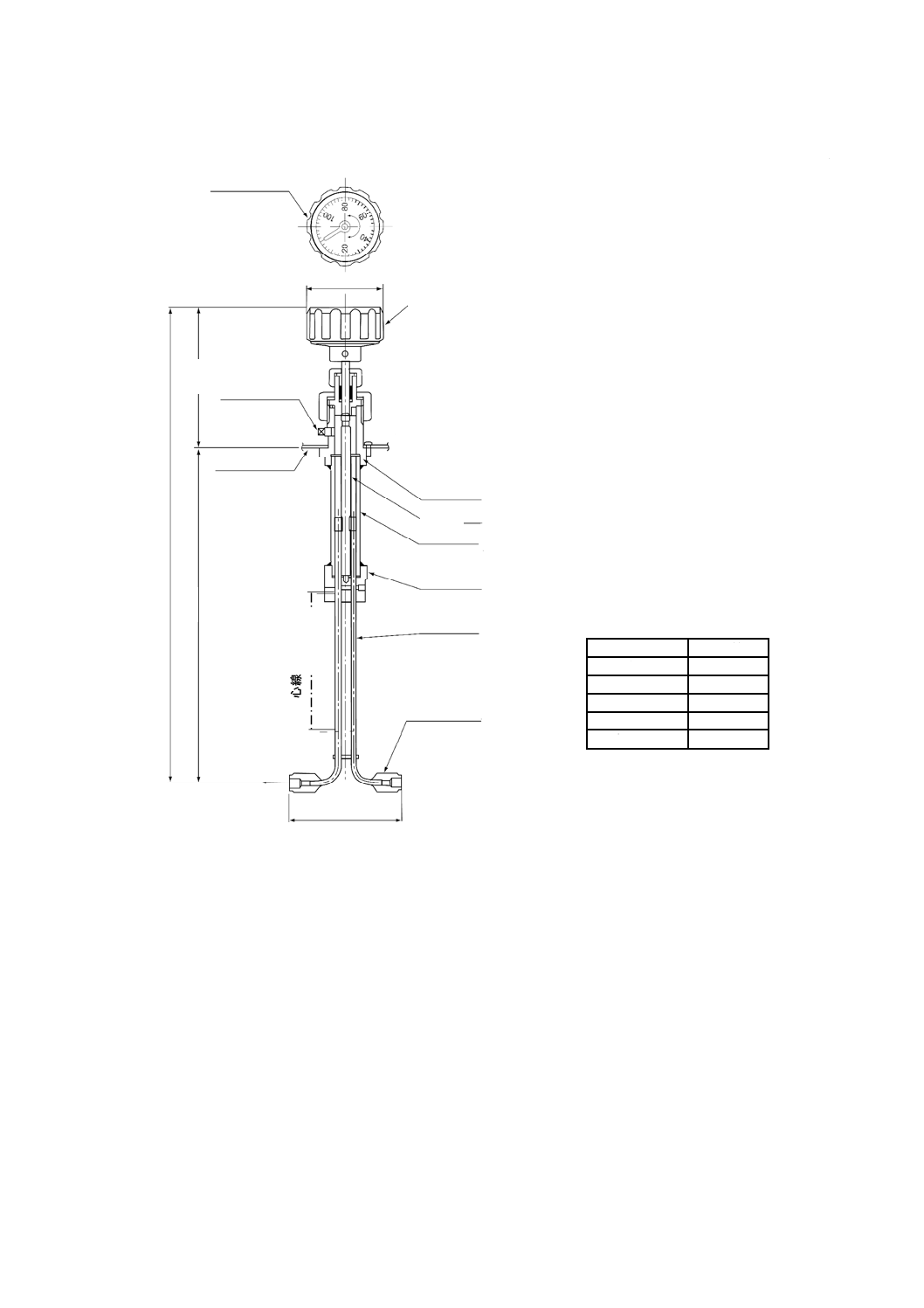
145
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
単位 mm
図A.7−二重心線式減圧器の一例
目盛板ストッパ
1
5
7
エアベントプラグ
取付板
指示計
スリーブ
ステム
パイプ
継手
チューブ
レデューサ
5
3
0
3
7
3
心
線
移
動
範
囲
130
主な構成部品の材質
部品名
材質
パイプ
SUS304TP
スリープ
SUS304
継手
SUS304
チューブ
SUS316TP
レデューサ
SUS304P
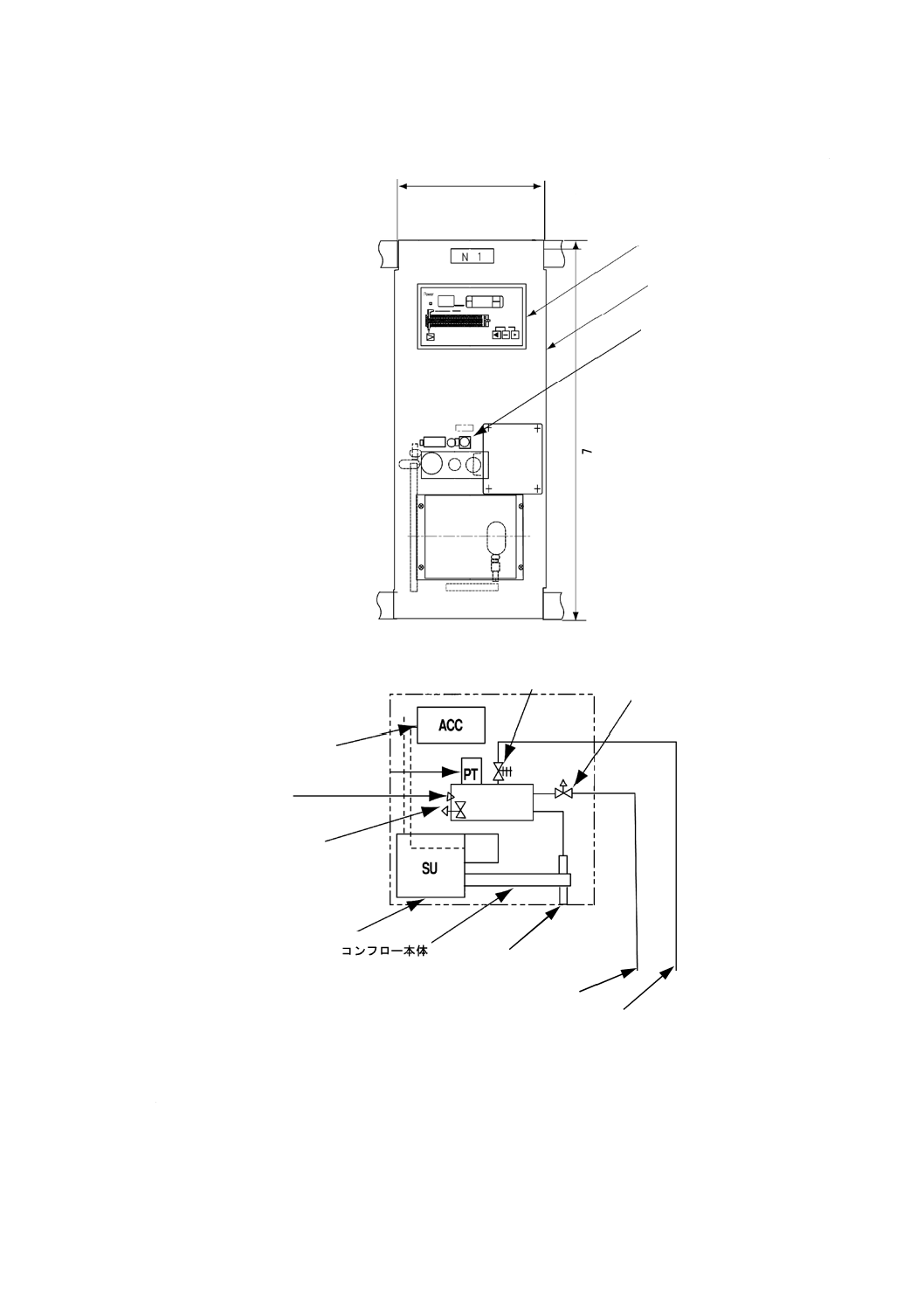
146
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
単位 mm
図A.8−自動減圧器の一例
4) ボイラ内試料採取管 ドラム形ボイラにおける連続ブロー装置又はボイラ水試料採取用内管の設置
位置及びその構造の代表的な例を,図A.9に示す。図A.9の中の配管用鋼管20 Aの長さは,ドラム
内に汽水分離器がある場合には,この汽水分離器の長さと等しくする。
280
コントローラ
パネル
ニードル弁
7
0
0
逃し弁
ニードル弁
コントローラ
圧力変換器
圧力変換器校正口
止め弁
サーボユニット コンフロー本体
試料水入口
試料水出口
逃し弁出口
147
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
単位 mm
図A.9−蒸気ドラム中の試料採取管の位置及び構造の一例
ボイラ水の試料は,ボイラ水の濃縮度が最も高い部分から採取するのが望ましいが,蒸発管から
の直接採取が困難なこともあって蒸発管で加熱されて上昇する濃縮水をドラムから採取するのが普
通である。降水管ではドラム内の水が給水によって希釈されているため,ドラム内の水より低い濃
度の試料を採取することになる。
なお,ドラムの内部には,給水管があり濃縮水が希釈される部位もあるので,試料採取用の内管
の取付位置については給水管との配置関係を十分に考慮しなければならない。すなわち,試料採取
用内管の吸込み孔は給水の吹出し方向には設けないようにしてある。図A.9に蒸気ドラム中の試料
採取管の位置及び構造の一例が示してある。また,ボイラ内試料採取管を取り付ける場合の設置条
件を満足させるものとして,連続ブロー用のボイラ水の採取内管を試料採取用内管の代用として使
用することができる。
5) 蒸気採取ノズル 蒸気採取ノズルの材料は,JIS G 3458で規定する配管用合金鋼鋼管を用い,その
設計基準は表A.1による。その構造の代表的な例を,図A.10に示す。
ノズルの取付位置は,一般に蒸気が蒸気管の内径の10倍以上の内径変化がない直管部を流れた位
置で,かつ,その位置の直後の至近距離に曲管部,内径変化,その他蒸気の流れを乱すような条件
がないところとする。
148
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表A.1−蒸気採取ノズルの設計基準
要目
蒸気管内径
D
(mm)
蒸気採取ノズルの蒸気取入れ孔
蒸気採取ノズル
の内径
g
(mm)
個数
n
配管位置
孔径
d
(mm)
r1
r2
r3
設計
基準
150以下
2
0.177 D 0.427 D
−
nE
S
D
06
.0
×
a)
n
d
5.1
×
以上
150を超える
3
0.144 D 0.337 D 0.454 D
注a) 蒸気採取ノズルの蒸気取入れ孔の孔径の算出式にあるSは,蒸気採取量(kg/min)であり,Eは蒸
気管内の蒸気流量(t/h)である。
単位 mm
図A.10−蒸気採取ノズルの構造の一例
蒸気の試料は,少量の湿分又は固体の不純物を含んでおり,一般には,これらの懸濁粒子を含む
流体の代表的試料を正しく適切に採取することは困難であるが,このような場合,複数の試料採取
孔をもつノズルを流体配管内に試料採取孔を上流側に向け,かつ,配管中心に向けてはめ合わせる
ことによって比較的正確に代表試料を採取することができる。
蒸気採取ノズルの設計に際しては,次の事項が重要である。
5.1) ノズル状の試料採取孔は,蒸気管の断面積を適切な数の環状の等面積に分割した各部を代表する
点に位置するようにする。
5.2) 等速吸引する。すなわち,採取する蒸気管内の蒸気流速と試料採取孔での試料の吸引速度を等し
くする。そのために試料採取孔の面積の合計と蒸気管断面積との比を,試料流量と蒸気流量との
比に等しくする必要がある。
これは,例えば,排ガス中のばいじんの測定の場合の試料の採取と同様である。
5.3) 各試料採取孔での吸引速度に差が生じないように,すなわち,各試料採取孔での圧力損失が等し
149
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
くなるようにする。このため,各試料採取孔の孔径d及び蒸気採取ノズルの内径gの設計に際し
ては,試料採取孔の面積の合計が,ノズルの内面積の2/3を超えないようにする。
5.1) の試料採取孔の数は,蒸気管の内径が大きくなるほど多くなるが,最大3(ASTM D 1066-82
による。)とした。また,試料採取孔の位置は,等面積の各環状区分の平均半径とした。以上の条
件を満たすようにした設計基準が表A.1である。
蒸気中の懸濁粒子の蒸気管断面積における分布は必ずしも均一ではなく,曲管部又は管の内径
変化及びその前後においては,分布の均一性を欠く傾向が強い。したがって,ノズルをはめ合わ
せる位置としては,蒸気管のなるべく長い直線部で,かつ,その前後に蒸気の流れを乱す曲管部
などがないところとした。
なお,はめ合わせるノズルは蒸気管径の全域にわたることが望ましいが,上述した取付け位置
を満足すれば,ノズルのはめ合わせる位置の蒸気管の断面における懸濁粒子の分布は,管軸を中
心にしてほぼ対象であると考えられるので,蒸気管内への挿入部はできる限り少なくし,更に強
度上の観点から短いほどよいことから,これらの事項を考慮して蒸気管の内径の1/2程度のノズル
を1本設置し,蒸気管の断面積の1/2に相当する部分から等速吸引して採取した試料を蒸気の代表
試料とすることとした。
蒸気採取ノズルの設計基準の計算例
設計条件 蒸気流量(E) 1 000 t/h
蒸気管内径(D) 350 mm
試料採取量(S) 2 kg/min
蒸気管の内径が150 mm以上であるので,表A.1から試料採取孔の数(n)は3となり,それぞ
れの孔の位置は次のようになる。
r1=0.144×D=50.4≒50
r2=0.337×D=117.95≒118
r3=0.454×D=158.9≒159
また,各採取孔の孔径(d)及び蒸気採取ノズルの内径(g)は,次のようになる。
mm
2.2
6
213
.2
000
1
3
2
06
.0
350
06
.0
≒
=
=
=
×
×
×
×
×
×
E
n
S
D
d
mm
7.4
9
666
.4
3
5.1
2.2
5.1
≒
=
=
=
×
×
×
×
n
d
g
蒸気採取ノズルの構造の代表例を図A.10に示すが,蒸気採取ノズルの肉厚,外径などは個々の
設計に際して十分に検討する必要があり,特に大口径の蒸気管の場合,及び蒸気条件が高圧,高
速の場合には,詳細な強度計算が必要である。
d) 試料採取操作
1) 試料採取装置の洗浄 試料採取装置の洗浄は,次による。
1.1) 試料採取装置を新設した場合 試料採取装置を新設した場合,ボイラプラントの各系統の試料導
管を取り付けた後,次の順序で洗浄を行う。
1.2) 試料導管部の洗浄 試料元弁を開いた後,試料導管フラッシング弁を開き,十分に試料導管部を
洗浄する。
1.3) 試料採取装置の洗浄 冷却器に冷却水を供給しながら化学分析系統だけに試料を流して,試料採
取装置内の洗浄を行う。次に,全系統に試料を24時間以上流して全体の洗浄を行う。
150
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1.4) ボイラ停止後再始動させる場合 ボイラを長期間停止し,再始動する場合には,試料導管に付着
している腐食生成物が流出するので,これらの影響を除くために新設時と同様の洗浄が必要であ
る。また,短時間停止し,再始動する場合には,試料導管,冷却器及び減圧装置中の滞留水を完
全に置換した後,更に滞留水の5倍量以上を流出させてから試料を採取する。
1.1) に準じて試料導管部及び試料採取装置の洗浄を行う。大形ボイラの場合には,ボイラが稼
働状態にあるときは,試料を設計試料採取流量で連続的に流しておく。
2) 採取操作 試料採取装置の試料出口管の端には軟質塩化ビニル管(ゴム管は用いない。)を取り付け
ておく。一般試験に用いる試料を採取する場合には,5.2.4 a) に従う。試料の採取量は,試験項目
数及び試験目的によって異なるが,一般には1〜2 Lでよい。
溶存酸素,ヒドラジニウムイオン及び亜硫酸イオンの試験に用いる試料の採取操作は,それぞれ
の試験項目で規定してあるのでそれに従う。
ヘキサン抽出物質を試験する試料を採取する場合には,一般の試験に用いる試料を採取する操作
とは異なるので注意する。すなわち,採取しようとする試料では試料容器を洗わずに,直接,試料
容器の底面に試料採取装置の試料出口管の端に取り付けた軟質ビニル管の先端が接触するようにし
て,試料容器の上に空間が残って(容量の約10 %)いる間に,軟質ビニル管を取り出して密栓する。
試料温度は,A.1.2 c) 2) に記載したように室温より約2 ℃低くなるようにするのが理想であるが,
一般の試験に用いる試料の場合には,室温又は室温より2〜3 ℃高くても特に支障はないが,溶存
酸素及び亜硫酸イオンを試験する試料の場合には,室温より約2 ℃低い温度とする。
3) 丸ボイラなどの低圧ボイラにおける試料採取方法 丸ボイラなどの低圧ボイラにおいては,一般に,
5.2.3 c) 4) で規定する試料採取管の設置がなく,ボイラ内処理剤の添加及びボイラ水のブローも間
欠的に行われている。したがって,ボイラ水の試験を行っても,得られた結果がボイラ水の水質を
代表していないことが多い。試料採取に当たっては,運転の状態をよく確認して試験の目的に対し
て最も適切な箇所を選んで採取する。
3.1) 採取時期 ボイラ水のブロー及び給水が間欠的に行われているボイラにおいては,ボイラ水が最
も濃縮された状態及び逆に最も濃縮されていない状態での試料についても試験して,水質の上限
及び下限値の管理を行う必要がある。
ボイラ内処理剤は,一般に,間欠的に使用されているが,ボイラ内処理剤を添加する前後に試
料を採取し,りん酸イオン,酸消費量などボイラ内処理剤によって影響を受ける項目について試
験を行い,その結果,著しい変動を示すようであれば,添加の間隔を短くして変動が少なくなる
ようにする。
3.2) 試料採取箇所 A.1.2 b) 2) に同じ。ただし,ボイラ水の水質の上限を管理する場合には,ボイラ
水の最も蒸発の盛んな場所,すなわち,最も濃縮された場所から採取する。この場合,ボイラ水
中に蒸気が混入しないような場所を選ぶ必要がある。また,水質の下限を管理する場合には,最
も濃縮されない場所,すなわち,ボイラの最下部から採取する。給水内管がボイラ底部及びボイ
ラの後部(横置ボイラの場合)に設置されている場合には,運転条件によってはボイラ底部と蒸
発面とでは塩類濃度が2倍以上も異なることがある。ボイラ底部の試料は,ボイラ底部のブロー
管に取り付けて採取する。
3.3) 採取方法 ボイラ水の試料を採取するとき冷却器を使用しないと,試料はフラッシュし,その組
成が変化するため試料としては不適切である。必ず冷却器を通して採取する。
完全な冷却器が設置できない場合には,蛇管だけを用意して,採取に当たってこれを,水を張
151
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
った容器中に浸して採取するのもよい。溶存酸素及び亜硫酸イオンを除く一般試験では,約60 ℃
以下まで冷却すれば大きな誤差は生じない。試料採取に当たっては少なくとも3分間以上試料を
流し,配管,冷却器内及び取出口付近のたまり水を流出させてから試料を採取する。
152
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書B
(参考)
濁度,アルカリ消費量,懸濁物質,100 ℃における過マンガン酸カリウム
による酸素消費量,溶存酸素,残留塩素,塩化物イオン,硫酸イオン,
りん酸イオン,アンモニア,ニッケル及びアルミニウムの測定に関する補足
B.1
濁度
B.1.1 一般事項
濁度は,水の濁りの程度を表すもので,視覚濁度,透過光濁度,散乱光濁度及び積分球濁度の四つの測
定法がある[JIS K 0101の9.(濁度)参照]。ボイラの給水・ボイラ水では,プラントの起動時に簡易的に
鉄濃度を把握するために測定し,主に,透過光濁度,散乱光濁度及び積分球濁度が用いられる。カオリン
標準液と比較して測定する場合には,“度(カオリン)”を単位とし,ホルマジン標準液と比較して測定す
る場合には,“度(ホルマジン)”を単位として表す。
濁度は変化しやすいので,試料採取後直ちに試験することが望ましい。
また,事前に濁度と鉄濃度との関係を把握しておくことが望ましい。
B.1.2 透過光濁度
試料を通過した波長660 nm付近の透過光の強度を測定し,カオリン標準液又はホルマジン標準液を用
いて作成した検量線から濁度を求める。
測定範囲:吸収セル50 mmのとき5〜50度(カオリン)又は4〜80度(ホルマジン)。
B.1.2.1 試薬
試薬は,次による。
a) 水 JIS K 0557で規定するA3の水を孔径約0.1 μmのろ過材を用いてろ過し,初めの約200 mLを捨
てた後のろ液。
b) 精製カオリン はくとう土(カオリン)約10 gをビーカ500 mLにとり,これに水300 mLとJIS K 8785
で規定する二りん酸ナトリウム十水和物0.2 gとを加え,マグネチックスターラで約3分間激しくかき
混ぜる。これをメスシリンダ(有栓形)1 000 mLに移し入れ,水を1 000 mLの標線まで加え,栓を
して約1分間激しく振り混ぜる。室温で約1時間静置した後,サイホンを用いて上部から250 mLの
液を捨て,次の500 mLまでの液を採取する。
採取した液を回転数約3 000 rpm(遠心分離器の回転部分の半径によって回転数を加減する。)で約
20分間遠心分離するか,又は孔径1 μm以下のろ過材によってろ過する。ろ別したカオリンを105〜
110 ℃で約3時間加熱し,デシケータ中で放冷した後,広口瓶に保存する。
c) カオリン標準液[1 000度(カオリン)] 精製カオリン1.00 gをとり適量の水に分散させた後,全量
フラスコ1 000 mLに移し入れ,水約800 mLとJIS K 8872で規定するホルムアルデヒド液約10 mLを
加えた後,水を標線まで加える。
d) カオリン標準液[100度(カオリン)] カオリン標準液[1 000度(カオリン)]をよく振り混ぜた後,
直ちにその100 mLを全量フラスコ1 000 mLにとり,水を標線まで加える。
e) ホルマジン標準液[400度(ホルマジン)] JIS K 8992で規定する硫酸ヒドラジニウム1.00 gをとり,
適量の水に溶かして,全量フラスコ100 mLに移し入れ,水を標線まで加える。
別に,JIS K 8847で規定するヘキサメチレンテトラミン10.0 gをとり,適量の水に溶かして,全量
153
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
フラスコ100 mLに移し入れ,水を標線まで加える。
この両溶液それぞれ10 mLを全量フラスコ200 mLにとり,よく振り混ぜる。液温25±3 ℃で約24
時間放置した後,水を標線まで加える。
B.1.2.2 器具及び装置
器具及び装置は,次による。
a) 光度計 分光光度計又は光電光度計
B.1.2.3 操作
操作は,次による。
a) カオリン標準液を用いる場合
1) 試料をよく振り混ぜた後,吸収セル50 mmにとり,波長660 nm付近における透過光の強度を見か
けの吸光度で測定する。
2) あらかじめ,次によって作成した検量線から試料の透過光濁度[度(カオリン)]を求める。
2.1) カオリン標準液[100度(カオリン)]5〜50 mLを全量フラスコ100 mLに段階的にとり,水を標
線まで加えて検量線用カオリン標準液[5〜50度(カオリン)]を調製する。1) の操作を行って検
量線用カオリン標準液の透過光濁度[度(カオリン)]と見かけの吸光度との関係線を作成する。
b) ホルマジン標準液を用いる場合
1) 試料をよく振り混ぜた後,a) 1) の操作を行う。
2) あらかじめ,次によって作成した検量線から試料の透過光濁度[度(ホルマジン)]を求める。
2.1) ホルマジン標準液[400度(ホルマジン)]1〜20 mLを全量フラスコ100 mLに段階的にとり,水
を標線まで加えて検量線用ホルマジン標準液[4〜80度(ホルマジン)]を調製する。1) の操作を
行って検量線用ホルマジン標準液の透過光濁度[度(ホルマジン)]と見かけの吸光度との関係線
を作成する。
B.1.2.4 留意事項
a) 吸収セル10 mmのとき,測定範囲は,25〜250度(カオリン)又は20〜400度(ホルマジン)となる。
b) 試料に色がある場合(特に,波長660 nm付近に吸収があるとき)は,試料を孔径1 μm以下のろ過材
を用いてろ過したろ液又は遠心分離した上澄み液を対照液にして,透過光の強度を見かけの吸光度と
して測定する。
B.1.3 散乱光濁度
試料中の粒子によって散乱した光の強度を波長660 nm付近で測定し,カオリン標準液又はホルマジン
標準液を用いて作成した検量線から求める。
測定範囲:1〜5度(カオリン)又は0.4〜5度(ホルマジン)(装置によって異なる。)
B.1.3.1 試薬
試薬は,次による。
a) 水 B.1.2.1 a) による。
b) カオリン標準液[100度(カオリン)] B.1.2.1 d) による。
c) ホルマジン標準液[40度(ホルマジン)] B.1.2.1 e) のホルマジン標準液[400度(ホルマジン)]10
mLを全量フラスコ100 mLにとり,水を標線まで加える。
B.1.3.2 器具及び装置
器具及び装置は,次による。
a) 散乱光濁度計 図B.1に散乱光濁度計の一例を示す。
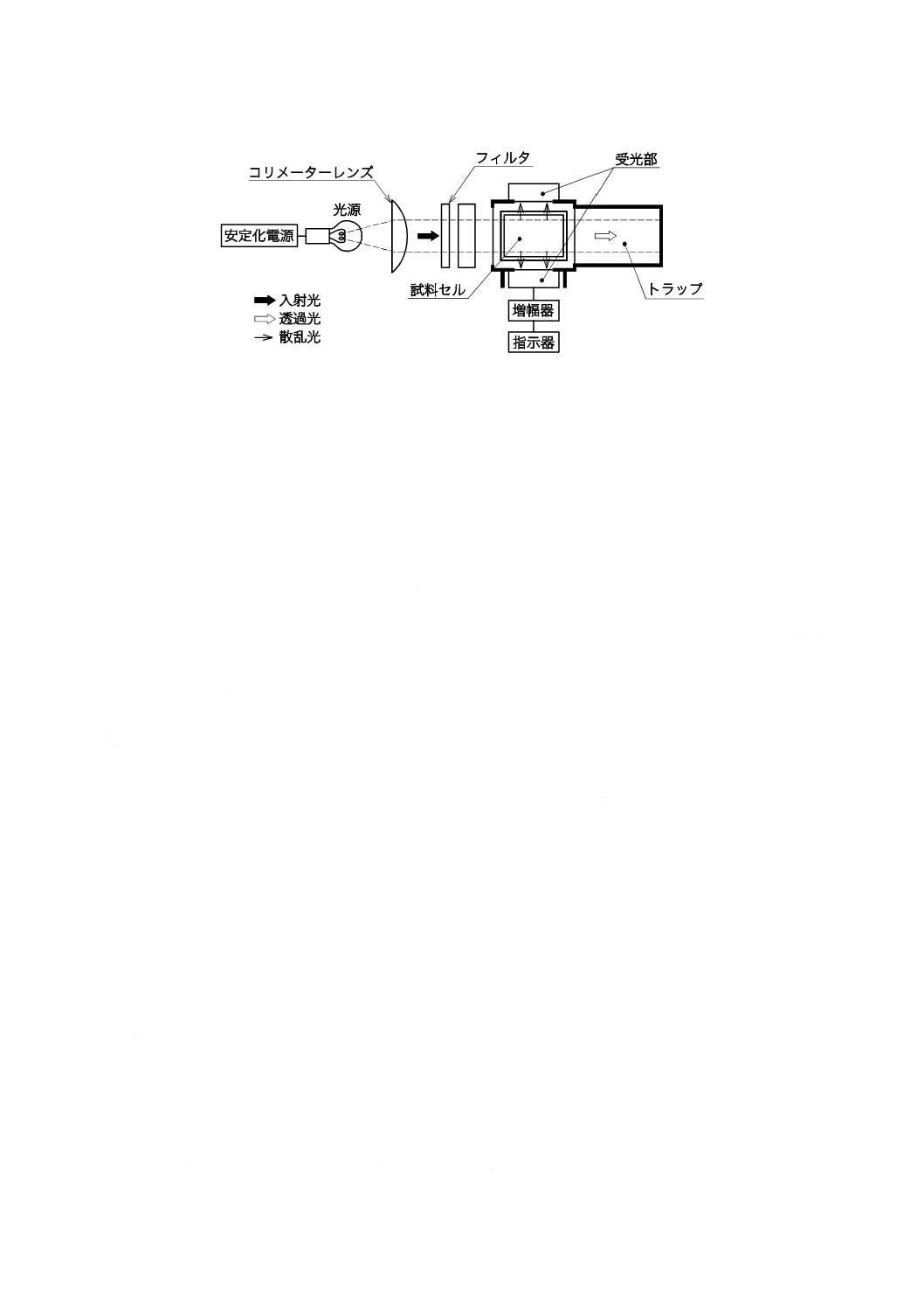
154
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図B.1−散乱光濁度計の構成例
B.1.3.3 操作
操作は,次による。
a) カオリン標準液を用いる場合
1) 水を吸収セルにとり,散乱光濁度計の指示値を0に調節し,次に,検量線用カオリン標準液[5度
(カオリン)]を用いて散乱光濁度計の指示値を100 %に調節する。
2) 試料をよく振り混ぜた後,吸収セルにとり,指示値(散乱光の強度)を測定する。
3) あらかじめ,次によって作成した検量線から試料の散乱光濁度[度(カオリン)]を求める。
3.1) カオリン標準液[100度(カオリン)]1〜5 mLを全量フラスコ100 mLに段階的にとり,水を標線
まで加えて検量線用カオリン標準液[1〜5度(カオリン)]を調製する。以下,1) 及び2) の操作
を行って検量線用カオリン標準液の散乱光濁度[1〜5度(カオリン)]と指示値(散乱光の強度)
との関係線を作成する。
b) ホルマジン標準液を用いる場合
1) 水を吸収セルにとり,散乱光濁度計の指示値を0に調節し,次に,検量線用ホルマジン標準液[5
度(ホルマジン)]を用いて散乱光濁度計の指示値を100 %に調節する。
2) 試料をよく振り混ぜた後,吸収セルにとり,指示値(散乱光の強度)を測定する。
3) あらかじめ,次によって作成した検量線から試料の散乱光濁度[度(ホルマジン)]を求める。
3.1) ホルマジン標準液[40度(ホルマジン)]1〜12.5 mLを全量フラスコ100 mLに段階的にとり,水
を標線まで加えて検量線用ホルマジン標準液[0.4〜5度(ホルマジン)]を調製する。以下,1) 及
び2) の操作を行って検量線用ホルマジン標準液の散乱光濁度[1〜5度(ホルマジン)]と指示値
(散乱光の強度)との関係線を作成する。
B.1.4 積分球濁度
水中の粒子による散乱光の強度と透過光の強度との比を求め,カオリン標準液又はホルマジン標準液を
用いて作成した検量線から求める。
測定範囲:吸収セル50 mmのとき0.2〜5度(カオリン)又は0.2〜5度(ホルマジン)
B.1.4.1 試薬
試薬は,次による。
a) 水 B.1.2.1 a) による。
b) カオリン標準液[100度(カオリン)] B.1.2.1 d) による。
c) ホルマジン標準液[40度(ホルマジン)] B.1.3.1 c) による。
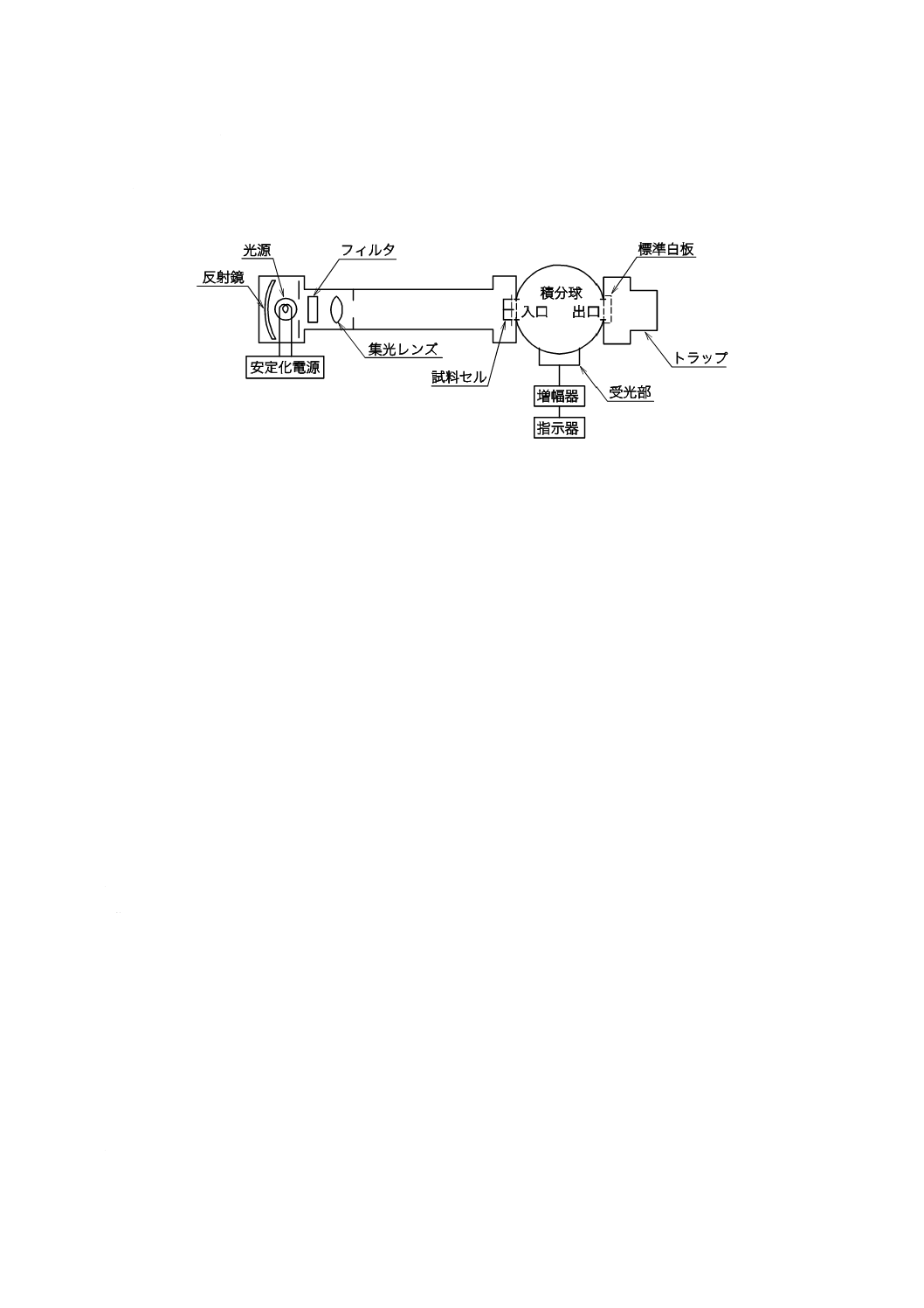
155
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
B.1.4.2 器具及び装置
器具及び装置は,次による。
a) 積分球濁度計 図B.2に積分球濁度計の一例を示す。
図B.2−積分球濁度計の構成例
B.1.4.3 操作
操作は,次による。
a) カオリン標準液を用いる場合
1) 水を入れた吸収セル50 mmとトラップを光路に入れて指示値を0に調節する。次に,トラップに標
準白板を挿入し,指示値が100になるように調節する。
2) 次に,水を入れた吸収セル50 mmに代えて,よく振り混ぜた試料を入れた吸収セル50 mmとトラ
ップ(標準白板は入れない)を光路に入れて散乱光の強度Tdを測定する。
3) 続いてトラップに標準白板を挿入して,試料の全透過光の強度Ttを測定する。
4) Td/Tt×100の値を算出し,あらかじめ,次によって作成した検量線から,試料の積分球濁度[度(カ
オリン)]を求める。
4.1) カオリン標準液[100度(カオリン)]0.2〜5 mLを全量フラスコ100 mLに段階的にとり,水を標
線まで加えて検量線用カオリン標準液[0.2〜5度(カオリン)]を調製する。1)〜3) の操作を行っ
て検量線用カオリン標準液の積分球濁度[度(カオリン)]とTd/Tt×100の値との関係線を作成す
る。
b) ホルマジン標準液を用いる場合
1) a) の1)〜3) の操作を行う。
2) Td/Tt×100の値を算出し,あらかじめ,次によって作成した検量線から試料の積分球濁度[度(ホ
ルマジン)]を求める。
2.1) ホルマジン標準液[40度(ホルマジン)]0.5〜12.5 mLを全量フラスコ100 mLに段階的にとり,
水を標線まで加えて検量線用ホルマジン標準液[0.2〜5度(ホルマジン)]を調製する。1) の操
作を行って検量線用標準液の積分球濁度[度(ホルマジン)]とTd/Tt×100の値との関係線を作成
する。
B.1.4.4 留意事項
a) 吸収セル10 mmのとき,測定範囲は,5〜100度(カオリン)又は5〜100度(ホルマジン)となる。
b) Td/Tt×100の計算を行わない場合は,次の方法を用いてもよい。

156
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
B.1.4.3 a) 1) の操作を行った後,水を入れた吸収セルに代えて試料を入れたセルを入れ,このとき
の指示値が100になるように調節する。続いて標準白板を外して指示値を読む。
検量線はカオリン標準液,又はホルマジン標準液について同様に操作し,横軸に積分球濁度[度(カ
オリン)],又は積分球濁度[度(ホルマジン)],縦軸に指示値(Td/Tt×100に相当する)をとって作
成し,この検量線から,試料の積分球濁度を求める。
B.1.5 濁度プロセス用分析装置による測定方法
測定範囲:0〜20度,20〜1 000度(ホルマジン),繰返し精度:最大目盛値の±3 %以下
B.1.5.1 測定原理
濁度プロセス用分析装置によって,給水及びボイラ水の濁度を連続的に測定する。
B.1.5.2 試薬
試薬は,次による。
a) 水 JIS K 0557で規定するA3の水。試薬の調製及び試験には,この水を用いる。
b) ホルマジン標準液[1 000度(ホルマジン)] JIS K 8992で規定する硫酸ヒドラジニウム1.25 gをと
り,水約200 mLに溶かす。別に,JIS K 8847で規定するヘキサメチレンテトラミン12.5 gをとり,
水約200 mLに溶かす。両者を全量フラスコ1 000 mLに移し入れ,よく振り混ぜた後,水を標線まで
加えて再びよく振り混ぜる。この溶液を25±3 ℃に保って24時間放置する。調製後30日間以上経過
したものは,使用しない。
c) ホルマジン標準液[100度(ホルマジン)] ホルマジン標準液[1 000度(ホルマジン)]100 mLを
全量フラスコ1 000 mLにとり,水を標線まで加える。この溶液は,使用時に調製する。
B.1.5.3 器具及び装置
器具及び装置は,次による。
a) 濁度プロセス用分析装置 箇条3 a) で規定するプロセス用分析装置で,JIS K 0801で規定する濁度自
動計測器の性能・機能を備えたもの。その構成例を,図B.3に示す。
図B.3−自動計測器の構成の一例
B.1.5.4 操作
操作は,次による。
a) 測定準備 濁度プロセス用分析装置の各部を点検し,特に,水漏れのないことを確認した後,所定の
手順に従って電源を入れ,各部が安定するまで待つ。
b) 校正 濁度プロセス用分析装置が安定状態に達した後,次の操作を行う。
1) ゼロ校正 B.1.5.2 a) の水を用いてゼロ校正を行う。または,電気的にゼロ校正を行う。
ゼロ校正は測定開始時に行う。
2) スパン校正 ホルマジン標準液[100度(ホルマジン)]を用いて測定レンジの25,50,75 %に相
当するスパン校正用のホルマジン標準液を調製してスパン校正を行う。
スパン校正は,測定開始時及び連続測定の場合は,1週間に1回程度の割合で行う。
157
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
c) 測定 校正が終了したら試料を設定流量で流し,指示値が安定した後,連続測定を行う。
B.1.5.5 点検・整備
点検・整備は,定期的に次のことを行うこととし,手順の詳細は製造業者の提供する取扱説明書による。
a) 試料の流量が所定量であることを確認する。
b) セル(検出部)の汚れがないことを確認する。
B.1.5.6 留意事項
a) 試験目的によって,低濃度,高濃度のレンジ切替えができる濁度プロセス用分析装置[例えば,0〜20
度(ホルマジン),20〜1 000度(ホルマジン)など]を用いるのが望ましい。
b) 装置に校正板が附属している場合には,校正板を光源と試料セルの間に挿入して,校正板の濁度を求
めておき,その後の日常のスパン校正には,これを用いてもよい。
B.2
アルカリ消費量
B.2.1 一般事項
アルカリ消費量は,水に溶けている強酸,炭酸,有機酸及び水酸化物として沈殿する金属元素などを所
定のpHまで中和するのに要する水酸化物イオンの量(アルカリの量)を試料1 Lについてのmmol数で表
すか,又は水酸化物イオンの量(アルカリの量)に相当する炭酸カルシウムの量に換算して,試料1 Lに
ついてのmg数で表す。
アルカリ消費量は,アルカリ消費量(pH 8.3),アルカリ消費量(pH 4.8)及びアルカリ消費量(遊離酸)
とに区分する。
アルカリ消費量は,JIS B 8223において,水質項目としての規定がない。
B.2.2 アルカリ消費量(pH 8.3)
アルカリ消費量(pH 8.3)は,pH計を用い,0.1 mol/L水酸化ナトリウム溶液で滴定して求める。
B.2.2.1 試薬
試薬は,次による。
a) 0.1 mol/L水酸化ナトリウム溶液 水約30 mLをポリエチレン瓶にとり,冷却しながらJIS K 8576で
規定する水酸化ナトリウム約30 gを少量ずつ加えて溶かし,密栓して4〜5日間放置する。その上澄
み液5 mLをポリエチレン製の気密容器1 Lにとり,4.7 a) の二酸化炭素を含まない水を加えて1 Lと
し,混合した後,二酸化炭素を遮断して保存する。この溶液は,次によって標定して用いる。
1) JIS K 8005で規定する容量分析用標準物質のアミド硫酸を上口デシケータ中に圧力2 kPa以下で約
48時間放置して乾燥する。その約0.2 gを1 mgの桁まではかりとり,三角フラスコ200 mLに入れ,
水約25 mLを加えて溶かす。
2) JIS K 8842で規定するブロモチモールブルー0.1 gをとり,JIS K 8102で規定するエタノール(95)
50 mLに溶かし,水で100 mLとしたブロモチモールブルー溶液(1 g/L)を,指示薬として1) に3
〜5滴を加え,この0.1 mol/L水酸化ナトリウム溶液で滴定し,溶液の色が緑になったときを終点と
する。次の式によって0.1 mol/L水酸化ナトリウム溶液のファクタ( f1)を算出する。
71
009
.0
1
100
1
×
×
×
=
x
b
a
f
ここに,
a: アミド硫酸の量(g)
b: アミド硫酸の純度(%)
x: 滴定に要した0.1 mol/L水酸化ナトリウム溶液(mL)
158
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
0.009 71: 0.1 mol/L水酸化ナトリウム溶液1 mLのアミド硫酸相当
量(g)
3) アミド硫酸による標定の代わりに,JIS K 8005で規定する容量分析用標準物質の炭酸ナトリウムを
用いて標定した0.1 mol/L塩酸を使用して,次のように標定してもよい。
0.1 mol/L塩酸20 mLをビーカにとり,指示薬として9.3.1 a) のフェノールフタレイン溶液(5 g/L)
3〜5滴を加えた後,この0.1 mol/L水酸化ナトリウム溶液で溶液が僅かに赤に呈色するまで滴定す
る。
滴定に要した0.1 mol/L水酸化ナトリウム溶液のmL数から,次の式によって0.1 mol/L水酸化ナ
トリウム溶液のファクタ( f1)を算出する。
x
f
f
2
1
20×
=
ここに,
f2: 0.1 mol/L塩酸のファクタ
x: 滴定に要した0.1 mol/L水酸化ナトリウム溶液(mL)
B.2.2.2 器具及び装置
器具及び装置は,次による。
a) pH計 JIS Z 8802の形式II
b) マグネチックスターラ
B.2.2.3 操作
操作は,次による。
a) 試料100 mLをビーカにとり,pH計の電極を入れる。
b) マグネチックスターラで緩やかにかき混ぜながら,0.1 mol/L水酸化ナトリウム溶液でpH 8.3になるま
で滴定する。必要な場合は,滴定曲線を作成して求める。pH計に代え,指示薬として9.3.1 a) のフェ
ノールフタレイン溶液(5 g/L)を用いて滴定してもよい。
c) 次の式によってアルカリ消費量(pH 8.3)を算出する。
mmol/Lで表す場合
V
f
a
A
000
1
1.0
1
×
×
×
=
ここに,
A: アルカリ消費量(pH 8.3)(mmol/L)
a: 滴定に要した0.1 mol/L水酸化ナトリウム溶液(mL)
f1: 0.1 mol/L水酸化ナトリウム溶液のファクタ
0.1: 0.1 mol/L水酸化ナトリウム溶液1 mLの水酸化物イオン
相当量(mmol)
V: 試料(mL)
CaCO3:mg/Lで表す場合
004
.5
000
1
1
×
×
×
=
V
f
a
B
ここに,
B: アルカリ消費量(pH 8.3)(CaCO3 mg/L)
a: 滴定に要した0.1 mol/L水酸化ナトリウム溶液(mL)
f1: 0.1 mol/L水酸化ナトリウム溶液のファクタ
V: 試料(mL)
5.004: 0.1 mol/L水酸化ナトリウム溶液1 mLの炭酸カルシウム
相当量(mg)
159
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
B.2.3 アルカリ消費量(pH 4.8)
アルカリ消費量(pH 4.8)は,pH計を用い,0.1 mol/L水酸化ナトリウム溶液で滴定して求める。
B.2.3.1 試薬
試薬は,次による。
a) 0.1 mol/L水酸化ナトリウム溶液 B.2.2.1 a) による。
B.2.3.2 器具及び装置
器具及び装置は,次による。
a) pH計 JIS Z 8802の形式II
b) マグネチックスターラ
B.2.3.3 操作
操作は,次による。
a) 試料100 mLをビーカにとり,pH計の電極を入れる。
b) マグネチックスターラで緩やかにかき混ぜながら,0.1 mol/L水酸化ナトリウム溶液でpH 4.8になるま
で滴定する。必要な場合は,滴定曲線を作成して求める。pH計に代え,指示薬として9.2.1 a) のメチ
ルレッド-ブロモクレゾールグリーン混合溶液を用いて滴定してもよい。
c) 次の式によってアルカリ消費量(pH 4.8)を算出する。
mmol/Lで表す場合
V
f
a
A
000
1
1.0
1
×
×
×
=
ここに,
A: アルカリ消費量(pH 4.8)(mmol/L)
a: 滴定に要した0.1 mol/L水酸化ナトリウム溶液(mL)
f1: 0.1 mol/L水酸化ナトリウム溶液のファクタ
0.1: 0.1 mol/L水酸化ナトリウム溶液1 mLの水酸化物イオン
相当量(mmol)
V: 試料(mL)
CaCO3:mg/Lで表す場合
004
.5
000
1
1
×
×
×
=
V
f
a
B
ここに,
B: アルカリ消費量(pH 4.8)(CaCO3:mg/L)
a: 滴定に要した0.1 mol/L水酸化ナトリウム溶液(mL)
f1: 0.1 mol/L水酸化ナトリウム溶液のファクタ
V: 試料(mL)
5.004: 0.1 mol/L水酸化ナトリウム溶液1 mLの炭酸カルシウム
相当量(mg)
B.2.4 アルカリ消費量(遊離酸)
アルカリ消費量(遊離酸)は,水に溶けている硫酸,塩酸,硝酸,強い有機酸などをしゅう酸カリウム
の存在下で0.1 mol/L水酸化ナトリウム溶液で滴定し,pH-水酸化ナトリウム溶液の滴定曲線から求める。
この測定値には,鉄,アルミニウムなどの塩類によるアルカリ消費量は含まれず,遊離酸に対応するア
ルカリ消費量が求まる。
B.2.4.1 試薬
試薬は,次による。
a) しゅう酸カリウム一水和物 JIS K 8522で規定するもの。
160
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) 0.1 mol/L水酸化ナトリウム溶液 B.2.2.1 a) による。
B.2.4.2 器具及び装置
器具及び装置は,次による。
a) pH計 JIS Z 8802の形式II
b) マグネチックスターラ
B.2.4.3 操作
操作は,次による。
a) 試料100 mLをビーカにとり,液温を約10 ℃に調節する。
b) しゅう酸カリウム一水和物約20 gを加え,かき混ぜて溶かす。
c) マグネチックスターラで緩やかにかき混ぜ,pH計を用いてpHを測定しながら0.1 mol/L水酸化ナト
リウム溶液で滴定する。
d) 滴定の終点(変曲点)前後では,0.1 mol/L水酸化ナトリウム溶液を0.1 mLずつ加える。
e) pHと0.1 mol/L水酸化ナトリウム溶液との滴定曲線を作成して終点を求め,次の式によってアルカリ
消費量(遊離酸)を算出する。
mmol/Lで表す場合
V
f
a
A
000
1
1.0
1
×
×
×
=
ここに,
A: アルカリ消費量(遊離酸)(mmol/L)
a: 滴定に要した0.1 mol/L水酸化ナトリウム溶液(mL)
f1: 0.1 mol/L水酸化ナトリウム溶液のファクタ
0.1: 0.1 mol/L水酸化ナトリウム溶液1 mLの水酸化物イオン
相当量(mmol)
V: 試料(mL)
CaCO3:mg/Lで表す場合
004
.5
000
1
1
×
×
×
=
V
f
a
B
ここに,
B: アルカリ消費量(遊離酸)(CaCO3:mg/L)
a: 滴定に要した0.1 mol/L水酸化ナトリウム溶液(mL)
f1: 0.1 mol/L水酸化ナトリウム溶液のファクタ
V: 試料(mL)
5.004: 0.1 mol/L水酸化ナトリウム溶液1 mLの炭酸カルシウム
相当量(mg)
B.3
懸濁物質,強熱残留物及び強熱減量
B.3.1 一般事項
水中に懸濁している物質及び水を蒸発したときの残留物質を懸濁物質,強熱残留物及び強熱減量に区分
して試験する。試験は,試料採取後,直ちに行う。直ちに行えない場合には,試料を0〜10 ℃の暗所に保
存し,できるだけ早く試験する。
懸濁物質,強熱残留物及び強熱減量は,JIS B 8223において,水質項目としての規定がない。
B.3.2 懸濁物質
試料をろ過し,ろ過材上に残留した物質を105〜110 ℃で加熱し,質量をはかる。
B.3.2.1 器具及び装置
161
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
器具及び装置は,次による。
a) ろ過器(分離形) 11.3.1 b) による。
b) ろ過材 11.3.1 c) による。
B.3.2.2 操作
操作は,次による。
a) ガラス繊維ろ紙を用いる場合は,あらかじめろ過器に取り付け,水で十分に吸引洗浄した後,このろ
過材を,時計皿上に置き,105〜110 ℃で約1時間加熱し,デシケータ中で放冷した後,その質量をは
かる。
注記 アクリル樹脂などのバインダ処理を行ったガラス繊維ろ紙,有機性ろ過膜,金属性ろ過膜は
洗浄しなくてよい。懸濁物質中の強熱残留物を定量する場合には,有機性ろ過膜を用いる。
有機性ろ過膜では105〜110 ℃で加熱すると変形するものがある。この場合は90 ℃で加熱す
る。時計皿はなるべく軽いものを用いるか,又はアルミニウムはくなどの軽い容器を用いる。
b) ろ過材をろ過器に取り付け,試料を十分に振り混ぜて懸濁物質が均一になってから,加熱後の懸濁物
質の量が2 mg以上になるように適量を手早く採取し,ろ過器に注ぎ入れて吸引ろ過する。試料容器
及びろ過管の器壁に付着した物質は,水でろ過材上に洗い落とし,ろ過材上の残留物質に合わせ,こ
れを水で数回洗浄する。
注記 ろ過しにくい試料の場合には,適量をビーカにとり,その都度よく振り混ぜて,液がろ過さ
れ終わる直前ごとに加え,ろ過速度が極めて遅くなったら試料の追加を止める。ビーカの中
の残量から試料の量を求める。
油脂,グリース,ワックスなどを含む試料で,これらを除いた懸濁物質を測定する場合に
は,試料をろ過した後,ろ過材を取り外すことなく,ろ過管ごと乾燥し,再び吸引瓶に取り
付けた後,JIS K 8848で規定するヘキサン10 mLずつを数回注ぎ入れ,油脂類を洗って除く。
c) 残留物は,ろ過材とともにピンセットなどを用いてろ過器から注意して取り外し,a) で用いた時計皿
上に移し,105〜110 ℃で2時間加熱し,デシケータ中で放冷した後,その質量をはかる。
d) 次の式によって懸濁物質(mg/L)を算出する。
V
b
a
S
000
1
)
(
×
−
=
ここに,
S: 懸濁物質(mg/L)
a: 懸濁物質を含んだろ過材及び時計皿の質量(mg)
b: ろ過材及び時計皿の質量(mg)
V: 試料(mL)
B.3.3 強熱残留物
B.3.2の懸濁物質,11.2の全蒸発残留物及び11.3の溶解性蒸発残留物のそれぞれを600±25 ℃で30分間
加熱したときの残留物で,それぞれの強熱残留物として示す。
B.3.3.1 懸濁物質の強熱残留物
B.3.3.1.1 試薬
試薬は,次による。
a) 硝酸アンモニウム溶液(250 g/L) JIS K 8545で規定する硝酸アンモニウム25 gを水に溶かして100
mLとする。
B.3.3.1.2 器具及び装置
162
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
器具及び装置は,次による。
a) 電気炉 600±25 ℃に調節できるもの。
b) るつぼ 白金又は磁器るつぼ10〜20 mL
B.3.3.1.3 操作
操作は,次による。
a) 600±25 ℃で加熱して恒量にしたるつぼにB.3.2.2 c) で得た懸濁物質を,ろ過材とともに移し入れ,
硝酸アンモニウム溶液(250 g/L)を滴加して湿した後,電気炉に入れ,徐々に温度を上げ600±25 ℃
で約30分間加熱して灰化し,デシケータ中で放冷した後,その質量をはかる。有機性ろ過膜がニトロ
化合物の場合には,JIS K 8839で規定する2-プロパノールを滴加して湿した後,強熱して灰化すると
よい。
b) 次の式によって強熱残留物(mg/L)を算出する。
なお,ろ過材にガラス繊維ろ紙を使用した場合には,あらかじめ別の同形のガラス繊維ろ紙を600
±25 ℃の電気炉で加熱して空試験値を求め,ガラス繊維ろ紙の質量を補正する。
V
b
a
R
000
1
)
(
×
−
=
ここに,
R: 懸濁物質の強熱残留物(mg/L)
a: 強熱残留物の入ったるつぼの質量(mg)
b: るつぼの質量(mg)
V: 試料(mL)
B.3.3.2 全蒸発残留物の強熱残留物
B.3.3.2.1 操作
11.2.2 c) で得た全蒸発残留物についてB.3.3.1.3の操作に準じて操作する。
B.3.3.3 溶解性蒸発残留物の強熱残留物
B.3.3.3.1 操作
11.3.2で得た溶解性蒸発残留物についてB.3.3.1.3の操作に準じて操作する。
B.3.4 強熱減量
B.3.3.1の懸濁物質の強熱残留物,B.3.3.2の全蒸発残留物の強熱残留物及びB.3.3.3の溶解性蒸発残留物
の強熱残留物の測定時における減少量で,それぞれの強熱減量として示す。
B.3.4.1 操作
懸濁物質の強熱減量,全蒸発残留物の強熱減量及び溶解性残留物の強熱減量は,次の式によって算出す
る。
E=S−R
ここに,
E: それぞれの強熱減量(mg/L)
S: それぞれの蒸発残留物(mg/L)
R: それぞれの強熱残留物(mg/L)
B.4
100 ℃における過マンガン酸カリウムによる酸素消費量(CODMn)
B.4.1 一般事項
試料を硫酸酸性とし,酸化剤として過マンガン酸カリウムを加え,沸騰水浴中で30分間反応させ,その
とき消費した過マンガン酸の量を求め,相当する酸素の量(O:mg/L)で表す。この試験は試料採取後,
直ちに行う。直ちに行えない場合には,0〜10 ℃の暗所に保存し,できるだけ早く試験する。
163
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
100 ℃における過マンガン酸カリウムによる酸素消費量(CODMn)は,JIS B 8223において,水質項目
としての規定がない。
定量範囲:O:0.5〜11 mg/L
B.4.2 試薬
試薬は,次による。
a) 水 JIS K 0557で規定するA4の水(CODMn値を与える物質を含んでいてはならない。)
b) 硫酸(1+2) 水2容をビーカにとり,これをかき混ぜながらJIS K 8951で規定する硫酸1容を徐々
に加えた後,僅かに赤に呈色するまで過マンガン酸カリウム溶液(3 g/L)を加える。
c) 硝酸銀溶液(200 g/L) JIS K 8550で規定する硝酸銀20 gを水に溶かして100 mLとする。着色ガラ
ス瓶に入れて保存する。
d) しゅう酸ナトリウム溶液(12.5 mmol/L) JIS K 8528で規定するしゅう酸ナトリウム1.8 gを水に溶
かして1 Lとする。ただし,e) の5 mmol/L過マンガン酸カリウム溶液のモル濃度の2.5倍より僅かに
高いモル濃度のものを調製する。
e) 5 mmol/L過マンガン酸カリウム溶液 JIS K 8247で規定する過マンガン酸カリウム0.8 gを平底フラ
スコにとり,水1 050〜1 100 mLを加えて溶かす。これを1〜2時間静かに煮沸した後,一夜放置する。
上澄み液をガラスろ過器G4を用いてろ過する(ろ過前後に水洗いしない。)。ろ液は,約30分間水蒸
気洗浄した着色ガラス瓶に入れて保存する。ファクタは,なるべく1に近い(0.95〜1.05)ものを使用
する。この溶液の標定は,次によって行う。
1) JIS K 8005で規定する容量分析用標準物質のしゅう酸ナトリウムをあらかじめ200 ℃で約1時間加
熱し,デシケータ中で放冷する。その約0.42 gを1 mgの桁まではかりとり,少量の水に溶かして,
全量フラスコ250 mLに移し入れ,水を標線まで加える。
2) この溶液25 mLを三角フラスコ300 mLにとり,水で約100 mLとし,硫酸(1+2)10 mLを加える。
液温25〜30 ℃で,ビュレットでこの5 mmol/L過マンガン酸カリウム溶液約22 mLを一度に加え,
赤い色が消えるまで放置する。
3) 次に,50〜60 ℃に加熱し,この5 mmol/L過マンガン酸カリウム溶液で滴定する。終点は僅かに赤
い色を約30秒間保つときとする。
4) 次の式によって,5 mmol/L過マンガン酸カリウム溶液のファクタ( f )を算出する。
675
001
.0
1
250
25
100
×
×
×
×
=
x
b
a
f
ここに,
a: しゅう酸ナトリウムの量(g)
b: しゅう酸ナトリウムの純度(%)
x: 滴定に要した5 mmol/L過マンガン酸カリウム溶液(mL)
0.001 675: 5 mmol/L過マンガン酸カリウム溶液1 mLのしゅう酸ナ
トリウム相当量(g)
B.4.3 器具及び装置
器具及び装置は,次による。
a) 水浴 試料を入れたとき,引き続いて沸騰状態を保てるような,熱容量及び加熱能力が大きなもの。
三角フラスコ300 mLが水浴の底に直接接触しないように,底から離して金網などを設ける。
B.4.4 操作
操作は,次による。
a) 試料100 mLを三角フラスコ300 mLにとり,硫酸(1+2)10 mLを加え,振り混ぜながら硝酸銀溶液
164
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(200 g/L)5 mLを加える。CODMnの濃度がO:11 mg/L以上の場合は,試料の適量をとり,水を加え
て100 mLとする。試料が懸濁物を含む場合には,よく振り混ぜて均一にした後,手早く採取する。
硝酸銀溶液(200 g/L)に代え,これに対応する硝酸銀の粉末を加えてもよい。
b) 5 mmol/L過マンガン酸カリウム溶液10 mLを加えて振り混ぜ,直ちに沸騰水浴中に入れ,30分間加
熱する。このとき,三角フラスコが倒れないように,その首に鉛製,鉄製などのリング状のおもりを
付ける。また,三角フラスコ300 mL中の試料の液面は,沸騰水浴の水面下になるように保つ。
c) 水浴から取り出し,しゅう酸ナトリウム溶液(12.5 mmol/L)10 mLを加えて振り混ぜ,よく反応させ
る。この際,塩化銀に酸化マンガン(IV)が混入し,反応にやや時間を要することがある。
d) 液温を50〜60 ℃で,5 mmol/L過マンガン酸カリウム溶液で僅かに赤に呈色するまで滴定する。
e) 別に,水100 mLを三角フラスコ300 mLにとり,a)〜d) の操作を行う。
f)
次の式によってCODMn(O:mg/L)を算出する。
(
)
2.0
000
1
Mn
×
×
×
−
=
V
f
b
a
COD
ここに,
CODMn: 100 ℃における過マンガン酸カリウムによる酸素消費量
(O:mg/L)
a: 滴定に要した5 mmol/L過マンガン酸カリウム溶液(mL)
b: 水を用いた試験の滴定に要した5 mmol/L過マンガン酸
カリウム溶液(mL)
f: 5 mmol/L過マンガン酸カリウム溶液のファクタ
V: 試料(mL)
0.2: 5 mmol/L過マンガン酸カリウム溶液1 mLの酸素相当量
(mg)
B.4.5 留意事項
a) 試料中に多量の塩化物イオンが存在する場合には当量になるまで硝酸銀溶液(200 g/L)を加え,更に
5 mL加える。
b) 塩化物イオンが多く,硝酸銀溶液(200 g/L)10 mL以上必要とする場合には,粉末にした硝酸銀を当
量よりも1 g過剰に加え,更に水5 mlを加える。
B.5
溶存酸素
B.5.1 一般事項
ほとんどの計器は温度補償,大気圧補償を自動で行い,また,入力した試料の塩分濃度を溶存酸素の濃
度計算に反映させる機能をもつ。このような自動機能をもたない計器を使用する場合は,使用者が温度及
び圧力などの影響を補正する必要がある。
なお,ボイラ給水及びボイラ水に含まれる塩分濃度は,極低濃度であるので塩分濃度の影響はないもの
とした。
B.5.2 標高及び気圧(例)
ある標高における実際の気圧を求める際は,表B.1の値を用いる。表B.1のデータは,標高0 mの大気
圧を1 013 hPaとして求めたものである。表B.1から得た気圧,又は地方気象台発表の大気圧を測定器に入
力する。
注記 計器が自動的に気圧補正を行う場合,この補正は不要である。
165
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表B.1−標高及び気圧(例)
標高
気圧
標高
気圧
m
hPa
m
hPa
0
1 013
1 800
815
150
995
1 950
800
300
979
2 100
785
450
960
2 250
771
600
943
2 400
756
750
926
2 550
742
900
910
2 700
728
1 050
893
2 850
715
1 200
877
3 000
701
1 350
861
3 150
688
1 500
846
3 300
675
1 650
830
−
−
B.5.3 飽和溶存酸素量と温度・気圧との関係
飽和溶存酸素量と温度・気圧との関係は,表B.2及び表B.3による。
表B.2−水への酸素溶解量と温度・気圧との関係(低気圧側のデータ)
温度
気圧 hPa
℃
733
767
800
833
867
900
933
酸素溶解量 mg/L
0
10.56
11.04
11.53
12.01
12.49
12.98
13.46
1
10.27
10.74
11.21
11.68
12.15
12.62
13.09
2
9.98
10.44
10.90
11.36
11.82
12.27
12.73
3
9.72
10.16
10.61
11.05
11.50
11.94
12.39
4
9.46
9.89
10.33
10.76
11.20
11.63
12.06
5
9.21
9.64
10.06
10.48
10.91
11.33
11.75
6
8.98
9.39
9.80
10.22
10.63
11.04
11.46
7
8.75
9.16
9.56
9.96
10.37
10.77
11.17
8
8.54
8.93
9.33
9.72
10.11
10.51
10.90
9
8.33
8.72
9.10
9.48
9.87
10.25
10.64
10
8.13
8.51
8.88
9.26
9.64
10.01
10.39
11
7.94
8.31
8.68
9.04
9.41
9.78
10.15
12
7.76
8.12
8.48
8.84
9.20
9.56
9.92
13
7.58
7.94
8.29
8.64
8.99
9.34
9.96
14
7.41
7.76
8.10
8.45
8.79
9.14
9.48
15
7.25
7.59
7.93
8.26
8.60
8.94
9.28
16
7.10
7.43
7.76
8.09
8.42
8.75
9.08
17
6.94
7.27
7.59
7.92
8.24
8.56
8.89
18
6.80
7.12
7.43
7.75
8.07
8.39
8.70
19
6.66
6.97
7.28
7.59
7.91
8.22
8.53
20
6.52
6.83
7.13
7.44
7.75
8.05
8.36
21
6.39
6.69
6.99
7.29
7.59
7.89
8.19
22
6.26
6.56
6.85
7.15
7.45
7.74
8.04
23
6.14
6.43
6.72
7.01
7.30
7.59
7.88
166
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表B.2−水への酸素溶解量と温度・気圧との関係(低気圧側のデータ)(続き)
温度
気圧 hPa
℃
733
767
800
833
867
900
933
酸素溶解量 mg/L
24
6.02
6.31
6.59
6.88
7.16
7.45
7.73
25
5.91
6.19
6.47
6.75
7.03
7.31
7.59
26
5.80
6.07
6.35
6.62
6.90
7.18
7.45
27
5.69
5.96
6.23
6.50
6.77
7.05
7.32
28
5.58
5.85
6.12
6.38
6.65
6.92
7.19
29
5.48
5.74
6.01
6.27
6.53
6.80
7.06
30
5.38
6.64
5.90
6.16
6.42
6.68
6.94
31
5.28
6.54
5.80
6.05
6.31
6.56
6.82
32
5.19
5.44
5.69
5.95
6.20
6.45
6.70
33
5.10
5.35
5.59
5.84
6.09
6.34
6.59
34
5.01
5.25
5.50
5.74
5.99
6.23
6.48
35
4.92
5.16
5.40
5.64
5.89
6.13
6.37
36
4.83
5.07
5.31
5.55
5.79
6.03
6.26
37
4.75
4.98
5.22
5.46
5.69
5.93
6.16
38
4.67
4.90
5.13
5.36
5.60
5.83
6.06
39
4.58
4.81
5.04
5.27
5.50
5.73
5.96
40
4.50
4.73
4.96
5.19
5.41
5.64
5.87
41
4.43
4.65
4.88
5.10
5.32
5.55
5.77
42
4.35
4.57
4.79
5.01
5.24
5.46
5.68
43
4.27
4.49
4.71
4.93
5.15
5.37
5.59
44
4.20
4.41
4.63
4.85
5.07
5.28
5.50
45
4.12
4.34
4.55
4.77
4.98
5.20
5.41
表B.3−水への酸素溶解量と温度・気圧との関係(高気圧側のデータ)
温度
気圧 hPa
℃
967
1 000
1 013
1 033
1 066
1 100
1 133
酸素溶解量 mg/L
0
13.94
14.43
14.62
14.91
15.39
15.88
16.36
1
13.56
14.03
14.22
14.50
14.97
15.44
15.91
2
13.19
13.65
13.83
14.10
14.56
15.02
15.48
3
12.84
13.28
13.46
13.73
14.17
14.62
15.06
4
12.50
12.93
13.11
13.37
13.80
14.24
14.67
5
12.18
12.60
12.77
13.02
13.45
13.87
14.29
6
11.87
12.28
12.45
12.69
13.11
13.52
13.93
7
11.57
11.98
12.14
12.38
12.78
13.19
13.59
8
11.29
11.69
11.84
12.08
12.47
12.87
13.26
9
11.02
11.41
11.56
11.79
12.17
12.56
12.94
10
10.76
11.14
11.29
11.51
11.89
12.26
12.64
11
10.51
10.88
11.03
11.25
11.61
11.98
12.35
12
10.27
10.63
10.78
10.99
11.35
11.71
12.07
13
10.04
10.40
10.54
10.75
11.10
11.45
11.80
14
9.82
10.17
10.31
10.51
10.86
11.20
11.54
15
9.61
9.95
10.08
10.29
10.62
10.96
11.30
16
9.41
9.74
9.87
10.07
10.40
10.73
11.06
167
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表B.3−水への酸素溶解量と温度・気圧との関係(高気圧側のデータ)(続き)
温度
気圧 hPa
℃
967
1 000
1 013
1 033
1 066
1 100
1 133
酸素溶解量 mg/L
17
9.21
9.54
9.67
9.86
10.18
10.51
10.83
18
9.02
9.34
9.47
9.66
9.98
10.29
10.61
19
8.84
9.15
9.28
9.46
9.77
10.09
10.40
20
8.66
8.97
9.09
9.28
9.58
9.89
10.19
21
8.49
8.79
8.92
9.10
9.40
9.70
10.00
22
8.33
8.63
8.74
8.92
9.21
9.51
9.80
23
8.17
8.46
8.58
8.75
9.04
9.33
9.62
24
8.02
8.30
8.42
8.59
8.87
9.16
9.44
25
7.87
8.15
8.26
8.43
8.71
8.99
9.27
26
7.73
8.00
8.11
8.28
8.55
8.83
9.11
27
7.59
7.86
7.97
8.13
8.40
8.67
8.94
28
7.45
7.72
7.83
7.99
8.25
8.52
8.79
29
7.32
7.59
7.69
7.85
8.11
8.37
8.64
30
7.20
7.46
7.56
7.71
7.97
8.23
8.49
31
7.07
7.33
7.43
7.58
7.84
8.09
8.35
32
6.95
7.20
7.31
7.46
7.71
7.96
8.21
33
6.84
7.08
7.18
7.33
7.58
7.83
8.08
34
6.72
6.97
7.07
7.21
7.46
7.70
7.95
35
6.61
6.85
6.95
7.09
7.34
7.58
7.82
36
6.50
6.74
6.84
6.98
7.22
7.46
7.70
37
6.40
6.63
6.73
6.87
7.10
7.34
7.57
38
6.29
6.53
6.62
6.76
6.99
7.22
7.46
39
6.19
6.42
6.52
6.65
6.88
7.11
7.34
40
6.09
6.32
6.41
6.55
6.78
7.00
7.23
41
6.00
6.22
6.31
6.45
6.67
6.90
7.12
42
5.90
6.12
6.21
6.35
6.57
6.79
7.01
43
5.81
6.03
6.12
6.25
6.47
6.69
6.91
B.5.4 水中の飽和溶存酸素に関する二つの算定方式
水中の飽和溶存酸素に関して,JIS B 8224:2005(以下,旧規格という。)の表17.2の数値は当時の陸水
学関係で一般的であったTruesdale et.al.,1955の1気圧での溶存酸素濃度の式を用いて飽和度を算定したも
のである。一方,この規格ではBenson et.al.,1984の圧力を考慮した溶存酸素濃度の式を用いて飽和度を算
定したものである。計算式が異なるため,両者の間には最大で3 %の差異がある。
この規格で採用しているBenson et.al.,1984の圧力を考慮した溶存酸素濃度から,旧規格で採用している
Truesdale et.al.,1955の1気圧での溶存酸素濃度へ換算する場合には,次の計算を行う。
表7で校正した溶存酸素計での実測値×{(旧規格の表17.2の飽和溶存酸素)/(表7の飽和溶存酸素)}
参考として,表B.4に旧規格の表17.2を示す。
168
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表B.4−水中の飽和溶存酸素(旧規格の表17.2参照)
水温
℃
溶存酸素
(O:mg/L)
水温
℃
溶存酸素
(O:mg/L)
水温
℃
溶存酸素
(O:mg/L)
水温
℃
溶存酸素
(O:mg/L)
0
14.16
11
10.67
22
8.53
33
7.23
1
13.77
12
10.43
23
8.39
34
7.13
2
13.40
13
10.20
24
8.25
35
7.04
3
13.04
14
9.97
25
8.11
−
−
4
12.70
15
9.76
26
7.99
−
−
5
12.37
16
9.56
27
7.87
−
−
6
12.06
17
9.37
28
7.75
−
−
7
11.75
18
9.18
29
7.64
−
−
8
11.47
19
9.01
30
7.53
−
−
9
11.19
20
8.84
31
7.43
−
−
10
10.92
21
8.68
32
7.32
−
−
B.6
残留塩素
B.6.1 一般事項
残留塩素とは,塩素剤が水に溶けて生成する次亜塩素酸及びこれがアンモニアと結合して生じるクロロ
アミンをいい,前者を遊離残留塩素,後者を結合残留塩素,両者を合わせて残留塩素という。
残留塩素の定量には,濃度の低い場合には,o-トリジン比色法又はジエチル-p-フェニレンジアミン(DPD)
比色法を適用し,濃度の高い場合には,よう素滴定法を適用する。この試験は,試料採取後,直ちに行う。
残留塩素は,JIS B 8223において,水質項目としての規定がない。
B.6.2 o-トリジン比色法
試料に3,3'-ジメチルベンジジン(o-トリジン)溶液を加え,残留塩素との反応で生じる黄色を,残留塩
素標準比色液と比較して残留塩素を定量する方法である。亜ひ酸ナトリウム溶液で処理し,残留塩素,遊
離残留塩素及び結合残留塩素の三つに区別することができる。
定量範囲:Cl:0.01〜2.0 mg/L,繰返し精度:5〜10 %
B.6.2.1 試薬
試薬は,次による。
a) 水 JIS K 0557で規定するA3の水(この試験に用いる水は,残留塩素のないこと及び塩素を消費し
ないこと。)
b) o-トリジン溶液 二塩化3,3'-ジメチルベンジジニウム(o-トリジン二塩酸塩)0.14 gを水50 mLに溶
かし,塩酸(3+7)(JIS K 8180で規定する塩酸を用いて調製する。)50 mL中にかき混ぜながら加え
る。着色瓶に入れて保存する。6か月以上経過したものは使用しない。
c) りん酸塩緩衝液(pH 6.5) JIS K 9020で規定するりん酸水素二ナトリウムを110 ℃で約2時間加熱
し,デシケータ中で放冷した後,その22.86 gと,JIS K 9007で規定するりん酸二水素カリウム46.14 g
とを水に溶かして1 Lとする。沈殿が生じた場合には,ろ別する。この溶液200 mLをとり水で1 Lと
する。
d) クロム酸カリウム-二クロム酸カリウム溶液 JIS K 8312で規定するクロム酸カリウム3.63 gとJIS K
8517で規定する二クロム酸カリウム1.21 gとをりん酸塩緩衝液(pH 6.5)に溶かし,全量フラスコ1 000
mLに移し入れ,りん酸塩緩衝液(pH 6.5)を標線まで加える。
e) 残留塩素標準比色液 相当する残留塩素の濃度(Cl:mg/L)に応じ,クロム酸カリウム-二クロム酸カ
169
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
リウム溶液及びりん酸塩緩衝液(pH 6.5)を表B.5に示す割合に比色管100 mLにとり,混ぜ合わせる。
暗所に保存する。沈殿が生じた場合には使用しない。
f)
亜ひ酸ナトリウム溶液(5 g/L) メタ亜ひ酸ナトリウム0.5 gを水に溶かして100 mLとする。
表B.5−残留塩素標準比色液(液層200 mm用)
残留塩素
Cl:mg/L
クロム酸カリウム-
二クロム酸カリウム溶液
mL
りん酸塩緩衝液
pH 6.5
mL
残留塩素
Cl:mg/L
クロム酸カリウム-
二クロム酸カリウム溶液
mL
りん酸塩緩衝液
pH 6.5
mL
0.01
0.18
99.82
0.70
7.48
92.52
0.02
0.28
99.72
0.80
8.54
91.46
0.05
0.61
99.39
0.90
9.60
90.40
0.07
0.82
99.18
1.00
10.66
89.34
0.10
1.13
98.87
1.10
12.22
87.78
0.15
1.66
98.34
1.20
13.35
86.65
0.20
2.19
97.81
1.30
14.48
85.52
0.25
2.72
97.28
1.40
15.60
84.40
0.30
3.25
96.75
1.50
16.75
83.25
0.35
3.78
96.22
1.60
17.84
82.16
0.40
4.31
95.69
1.70
18.97
81.03
0.45
4.84
95.16
1.80
20.09
79.91
0.50
5.37
94.63
1.90
21.22
78.78
0.60
6.42
93.58
2.00
22.34
77.66
B.6.2.2 器具及び装置
器具及び装置は,次による。
a) 比色管 100 mL 底部から200±1.5 mmの高さに100 mLの標線を付けた平底のもの。
b) 比色管立 100 mL用 底部及び側面に乳白板を付けたもの。
B.6.2.3 操作
操作は,次による。
a) 比色管にo-トリジン溶液5 mLをとり,これに試料の適量(残留塩素2.0 mg以下を含む。)を加え,更
に水を100 mLの標線まで加え,手早く栓をして振り混ぜる。
なお,試料がアルカリ性の場合には,pH計を用い,塩酸(1+5)を加えてpHを約7にする。また,
発色時のpHは常に1.3以下とする。
b) 5分間暗所に放置する。
注記 残留塩素のうち結合残留塩素は,最高発色に達するのに0 ℃で6分間,20 ℃で3分間,25 ℃
で2分30秒間が必要である。
c) 上方から透視して残留塩素標準比色液と比較し,該当する残留塩素標準比色液を求め,これに相当す
る残留塩素の濃度a(Cl:mg/L)を記録する。
d) 別の比色管にo-トリジン溶液5 mLをとり,これにa) の操作と同量の試料を加え,手早く栓をして振
り混ぜる。
e) 5秒間以内に亜ひ酸ナトリウム溶液(5 g/L)5 mLを加えて振り混ぜ,更に,水を100 mLの標線まで
加えて振り混ぜる。
f)
残留塩素標準比色液と比較し,該当する残留塩素標準比色液を求め,これに相当する残留塩素の濃度
170
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b(Cl:mg/L)を記録する。
g) 空試験として比色管100 mLに亜ひ酸ナトリウム溶液(5 g/L)5 mLをとり,これにa) の操作と同量
の試料を加えて振り混ぜる。
h) o-トリジン溶液5 mLを加えて振り混ぜ,更に,水を100 mLの標線まで加えて振り混ぜる。
i)
5秒間以内に残留塩素標準比色液と比較し,該当する残留塩素標準比色液を求め,これに相当する残
留塩素の濃度c1(Cl:mg/L)を記録する。
j)
さらに,5分間暗所に放置後,残留塩素標準比色液と比較し,該当する残留塩素標準比色液を求め,
これに相当する残留塩素の濃度c2(Cl:mg/L)を記録する。
k) 次の式によって残留塩素,遊離残留塩素及び結合残留塩素の濃度を算出する。
(
)
V
c
a
A
100
2×
−
=
(
)
V
c
b
B
100
1×
−
=
B
A
C
−
=
ここに,
A: 残留塩素(Cl:mg/L)
a: c) で求めた残留塩素(Cl:mg/L)
c2: j) で求めた残留塩素(Cl:mg/L)
V: 試料(mL)
B: 遊離残留塩素(Cl:mg/L)
b: f) で求めた残留塩素(Cl:mg/L)
c1: i) で求めた残留塩素(Cl:mg/L)
C: 結合残留塩素(Cl:mg/L)
B.6.2.4 留意事項
a) 空試験を行わない場合には,鉄0.3 mg/L以上,マンガン10 μg/L以上又は亜硝酸イオン0.3 mg/L以上
が含まれていると妨害する。鉄及びマンガンの妨害を防ぐには,試料100 mLにつき1,2-シクロヘキサ
ンジアミン四酢酸溶液(10 g/L)(trans-1,2-シクロヘキサンジアミン四酢酸一水和物1.05 gを水に溶か
して100 mLとする。)3 mLを添加する。
b) 市販の残留塩素測定器を用いる場合には,あらかじめ残留塩素標準比色液と比較して,誤りのないこ
とを確認しておく。
警告 o-トリジンはIARCで2B(ヒト発がん性の可能性がある物質),日本産業衛生学会で2B(人
間に対しておそらく発がん性があると考えられる物質・証拠が比較的十分でない物質)に分
類されている。また,メタ亜ひ酸ナトリウムは毒物及び劇物取締法の毒物“砒素化合物”に
分類されており,発がん性についてもIARCで1(発がん性の十分なデータがある物質)に
分類されている。そのため,取扱いに当たっては法令に従い安全及び健康に対する適切な措
置をとらなければならない。また,廃液の処分には特に注意する。
B.6.3 ジエチル-p-フェニレンジアミン(DPD)比色法
硫酸N,N-ジエチル-p-フェニレンジアンモニウム(DPD)を比色管にとり,これに試料を加え,残留塩素
との反応で生じる桃色から桃紅色を,残留塩素標準比色液と比較して定量する。
定量範囲:Cl:0.05〜2 mg/L,繰返し精度:5〜10 %
B.6.3.1 試薬
試薬は,次による。

171
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) 水 B.6.2.1 a) による。
b) よう化カリウム JIS K 8913で規定するもの。
c) DPD希釈粉末 硫酸N,N-ジエチル-p-フェニレンジアンモニウム(N,N-ジエチル-p-フェニレンジアミ
ン硫酸塩)1.0 gをめのう乳鉢中で粉砕する。これにJIS K 8987で規定する硫酸ナトリウム24 gを加
えてよく混合し,着色ガラス瓶に入れ,湿気を避けて,0〜10 ℃の暗所に保存する。着色したものは
使用しない。
d) りん酸塩緩衝液(pH 6.5) りん酸二水素カリウム溶液(0.2 mol/L)(JIS K 9007で規定するりん酸二
水素カリウム27.2 gを水に溶かして1 Lとする。)100 mLをとり,水酸化ナトリウム溶液(0.2 mol/L)
(JIS K 8576で規定する水酸化ナトリウム8 gを水に溶かして1 Lとする。)を,pH計を用いてpH 6.5
になるまで加え,これにtrans-1,2-シクロヘキサンジアミン四酢酸一水和物0.13 gを加えて溶かす。
e) C. I. Acid Red 265溶液 C. I. Acid Red 265[1-(4-メチルベンゼンスルホンアミド)-7-(2-メチルフェ
ニルアゾ)-8-ヒドロキシ-3,6-ナフタレンジスルホン酸二ナトリウム]を105〜110 ℃で3〜4時間加熱
し,デシケータ中で放冷する。その0.329 gを1 mgの桁まではかりとり,少量の水に溶かして全量フ
ラスコ1 000 mLに移し入れ,水を標線まで加える。この溶液50 mLを全量フラスコ500 mLにとり,
水を標線まで加える。0〜10 ℃の暗所に保存し,6か月間以上経過したものは使用しない。
f)
DPD残留塩素標準比色液 C. I. Acid Red 265溶液を表B.6によって全量フラスコ50 mLにとり,水を
標線まで加える。これを比色管50 mLにそれぞれ移す。密栓して0〜10 ℃の暗所に保存する。6か月
間以上経過したものは使用しない。
表B.6−DPD残留塩素標準比色液(50 mL中)
残留塩素
Cl:mg/L
C. I. Acid Red 265溶液
mL
0.05
0.5
0.1
1.0
0.2
2.0
0.3
3.0
0.4
4.0
0.5
5.0
0.6
6.0
0.7
7.0
0.8
8.0
0.9
9.0
1.0
10.0
1.2
12.0
1.4
14.0
1.6
16.0
1.8
18.0
2.0
20.0
B.6.3.2 器具及び装置
器具及び装置は,次による。
a) 比色管 50 mL 底部から150±1 mmの高さに50 mLの標線を付けた平底のもの。
b) 比色管立 50 mL用 底部及び側面に乳白板を付けたもの。
172
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
B.6.3.3 操作
操作は,次による。
a) りん酸塩緩衝液(pH 6.5)2.5 mLを比色管50 mLにとり,これにDPD希釈粉末0.5 gを加える。次に,
試料の適量(残留塩素2.0 mg/L以下を含む。)を加え,更に水を標線まで加える。
なお,試料の酸性又はアルカリ性が強い場合には,炭酸ナトリウム溶液(50 g/L)(JIS K 8625で規
定する炭酸ナトリウムを用いて調製する。)又は塩酸(1+11)[8.3.1 a) による。]を用いてpHを約
6.5に調節する。
b) 栓をしてよく振り混ぜ,1分間以内にその発色を側面から透視して,DPD残留塩素標準比色液と比較
する。該当するDPD残留塩素標準比色液から,これに相当する残留塩素の濃度(Cl:mg/L)を求め,
これを遊離残留塩素として,試料中の遊離残留塩素の濃度(Cl:mg/L)を算出する。反応時間の計測
には振り混ぜ時間を含める。DPD希釈粉末中の硫酸ナトリウムは完全には溶けなくてもよい。
c) b) の操作が終了したら,よう化カリウム約0.5 gを加え,栓をして振り混ぜて溶かし,約2分間放置
後,その発色をb) と同様に残留塩素標準比色液と比較する。該当する残留塩素標準比色液からこれ
に相当する残留塩素の濃度(Cl:mg/L)を求め,試料中の残留塩素の濃度を算出する。
d) 結合残留塩素の濃度(Cl:mg/L)は,次の式によって算出する。
結合残留塩素の濃度(Cl:mg/L)=残留塩素(Cl:mg/L)−遊離残留塩素(Cl:mg/L)
B.6.3.4 留意事項
a) 市販の残留塩素測定器又はガラス色標準スケールを用いる場合には,あらかじめDPD残留塩素標準比
色液と比較して,誤りのないことを確認しておく。
b) 試料中のマンガン酸化物による妨害の補正方法は,次による。
1) 試料100 mLをとり,メタ亜ひ酸ナトリウム溶液(2 g/L)(メタ亜ひ酸ナトリウムを用いて調製する。)
又はチオアセトアミド(エタンチオアミド)溶液(2.5 g/L)1 mLを加え,振り混ぜた後,りん酸塩
緩衝液(pH 6.5)2.5 mL及びDPD希釈粉末0.5 gを加え,振り混ぜる。
2) この溶液を用いて,B.6.3.3のb) 及びc) の操作を行い,マンガン酸化物による発色を残留塩素の濃
度として求める。
3) 得られた値を用いて,遊離残留塩素の濃度及び残留塩素の濃度を補正する。
c) 二酸化塩素(IV)は,残留塩素及び遊離残留塩素の値に含まれる。
d) DPDの酸化は,塩素化合物によるものだけではない。反応は,他の酸化剤によっても生じる。これに
は臭素,よう素,ブロモアミン類,ヨードアミン類,オゾン,過酸化水素,クロム酸塩,マンガン酸
化物,亜硝酸塩,鉄(III)イオン及び銅(II)イオンが挙げられる。ただし,この方法では,銅(II)
イオン2 mg/L,鉄(II)イオン3 mg/L,アルミニウムイオン4 mg/L及び亜硝酸イオン4 mg/Lまでは
それぞれ妨害しない。
B.6.4 よう素滴定法
残留塩素とよう化カリウムとが反応して遊離するよう素をチオ硫酸ナトリウム溶液で滴定し,残留塩素
を定量する。よう素を遊離させる酸化性物質が共存すると,残留塩素として定量される。
定量範囲:Cl:0.1 mg/L以上
B.6.4.1 試薬
試薬は,次による。
a) 水 B.6.2.1 a) による。
b) よう化カリウム JIS K 8913で規定するもの。
173
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
c) 酢酸(1+1) JIS K 8355で規定する酢酸を用いて調製する。
d) でんぷん溶液(10 g/L) 16.2.1 g) による。
e) 10 mmol/Lチオ硫酸ナトリウム溶液 16.2.1 k) による。
B.6.4.2 操作
操作は,次による。
a) 試料の100 mLを共栓三角フラスコ500 mLにとり,水を加えて約300 mLとし,よう化カリウム1 g
及び酢酸(1+1)5 mLを加える。残留塩素濃度が70 mg/L以上の場合は,試料の適量をとる。
なお,試料がアルカリ性の場合には,pH計を用い,塩酸(1+5)を加えてpHを約7にする。
b) 栓をして振り混ぜ,暗所に約5分間放置する。
c) 遊離したよう素を,10 mmol/Lチオ硫酸ナトリウム溶液で滴定し,溶液の黄色が薄くなってから,指
示薬としてでんぷん溶液(10 g/L)1 mLを加え,生じたよう素でんぷんの青い色が消えるまで滴定す
る。
d) 空試験として水100 mLをとり,a)〜c) の操作を行う。
e) 次の式によって試料中の残留塩素の濃度(Cl:mg/L)を算出する。
(
)
5
354
.0
000
1
×
×
×
−
=
V
f
b
a
A
ここに,
A: 残留塩素(Cl:mg/L)
a: 滴定に要した10 mmol/Lチオ硫酸ナトリウム溶液(mL)
b: 空試験に要した10 mmol/Lチオ硫酸ナトリウム溶液
(mL)
f: 10 mmol/Lチオ硫酸ナトリウム溶液のファクタ
V: 試料(mL)
0.354 5: 10 mmol/Lチオ硫酸ナトリウム溶液1 mLの残留塩素相
当量(mg)
B.7
塩化物イオン(Cl−)
B.7.1 一般事項
硝酸水銀(II)の試薬が,有害であり試薬,廃液とも取扱いが難しいため,塩化銀比濁法を採用した上
で,硝酸水銀(II)滴定法は,この附属書(参考)に記載する。
B.7.2 硝酸水銀(II)滴定法
試料のpHを2.5に調節し,硝酸水銀(II)溶液で滴定して塩化物イオンを定量する。よう化物イオン及
び臭化物イオンは塩化物イオンとして定量される。亜硫酸イオン,ヒドラジニウムイオン,ヒドロキシル
アミンなどの還元性物質は妨害するが,過酸化水素で酸化すれば妨害しない。クロム(VI)及び鉄(III)
は,いずれも10 mg/L以下であれば妨害しない。
定量範囲:Cl−:1 mg/L以上
B.7.2.1 試薬
試薬は,次による。
a) 水 JIS K 0557で規定するA3又はA4の水
b) 硝酸(1+65) JIS K 8541で規定する硝酸を用いて調製する。
c) 過酸化水素(1+1) JIS K 8230で規定する過酸化水素を用いて調製する。
d) 混合指示薬 ジフェニルカルバゾン0.50 g,JIS K 8844で規定するブロモフェノールブルー0.05 g及び
174
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
JIS K 8272で規定するキシレンシアノールFF 0.12 gをはかりとり,JIS K 8102で規定するエタノール
(95)100 mLに溶かし,着色ガラス瓶に入れ保存する。
e) 塩化物イオン標準液(Cl−:500 mg/L) 15.2.1 d) の塩化物イオン標準液(Cl−:1 000 mg/L)100 mL
を全量フラスコ200 mLにとり,水を標線まで加える。
f)
7.05 mmol/L硝酸水銀(II)溶液 硝酸水銀(II)n水和物2.5 gをとり,JIS K 8541で規定する硝酸
0.5 mLを含む水20 mLを加えて溶かし,ビーカ1 000 mLに移し入れ,水で1 Lとする。
標定 塩化物イオン標準液(Cl−:500 mg/L)20 mLをビーカにとり,水を加えて100 mLとし,混合
指示薬5滴を加え,溶液の色が青から青緑になるまで硝酸(1+65)を滴加し,更に1 mLを加
えpHを約2.5にする。次に,この7.05 mmol/L硝酸水銀(II)溶液で滴定し,溶液の色が黄緑
から灰色を経て紫になるときを終点とする。別に,水100 mLをとり,空試験を行って滴定値
を補正する。補正したmL数(x)から,次の式によって7.05 mmol/L硝酸水銀(II)溶液のフ
ァクタ( f )を算出する。
x
f
20
=
B.7.2.2 操作
操作は,次による。
a) 試料に濁りがある場合にはろ紙5種Cでろ過し,初めのろ液約50 mLを捨て,次のろ液100 mL(Cl −:
50 mg/L以上を含む場合には,適量をとり,水を加えて100 mLとする。)をビーカにとる。ただし,
試料に著しい濁りが認められない場合は,ろ過操作を省略してもよい。
b) 亜硫酸イオン,ヒドラジニウムイオン,ヒドロキシルアミンなどの還元性物質が共存する場合には,
過酸化水素(1+1)を滴加し,かき混ぜて分解する。
c) 混合指示薬5滴を加え,溶液の色が明らかに青から青緑又は黄緑になるまで硝酸(1+65)を滴加した
後,更に1 mLを加えて,pHを約2.5にする。
なお,酸性が強い場合には,水酸化ナトリウム溶液(40 g/L)でpH約2.5に調節する。
d) 7.05 mmol/L硝酸水銀(II)溶液で滴定し,溶液の色が黄緑から灰色を経て紫になるときを終点とする。
e) 空試験として水100 mLをとり,c) 及びd) の操作を行う。
f)
次の式によって試料中の塩化物イオンの濃度(Cl−:mg/L)を算出する。
5.0
000
1
)
(
×
×
×
V
f
b
a
C
−
=
ここに,
C: 塩化物イオン(Cl−:mg/L)
a: 滴定に要した7.05 mmol/L硝酸水銀(II)溶液(mL)
b: 空試験の滴定に要した7.05 mmol/L硝酸水銀(II)溶液
(mL)
f: 7.05 mmol/L硝酸水銀(II)溶液のファクタ
V: 試料(mL)
0.5: 7.05 mmol/L硝酸水銀(II)溶液1 mLの塩化物イオン相
当量(mg)
警告 水銀化合物を使用するため,法令に従い安全及び健康に対する適切な措置を取らなければなら
ない。また,廃液の処分には特に注意する。
175
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
B.8
硫酸イオン(SO42−)
B.8.1 一般事項
クロム酸バリウム−ジフェニルカルバジド吸光光度法及びクロム酸バリウム吸光光度法は操作が煩雑で
適用頻度が少ないため,この附属書(参考)に記載する。
B.8.2 クロム酸バリウム−ジフェニルカルバジド吸光光度法
試料にクロム酸バリウムの酸懸濁液を加えて硫酸バリウムを沈殿させ,次に,カルシウムイオンを含む
アンモニア水とエタノールとを加え,過剰のクロム酸バリウムを沈殿させ,遠心分離する。硫酸イオンと
置換して生じたクロム酸イオンを二クロム酸イオンに変え,これとジフェニルカルバジド(1,5-ジフェニ
ルカルボノヒドラジド)とを反応させ,生じる赤紫の吸光度を測定して硫酸イオンを定量する。
定量範囲:SO42− 0.2〜5 mg/L,繰返し精度:3〜10 %
B.8.2.1 試薬
試薬は,次による。
a) 塩酸(1+1) 21.5.1 b) による。
b) クロム酸バリウムの酸懸濁液(A) クロム酸バリウム0.5 gを酢酸(1+15)100 mLと塩酸(1+500)
100 mLとの混合溶液200 mLに加え,よく振り混ぜて懸濁液を作り,ポリエチレン瓶に保存する。ク
ロム酸バリウムは,次の方法で調製する。JIS K 8312で規定するクロム酸カリウム8 gを水約800 mL
に溶かし,酢酸(6 moL/L)(JIS K 8355で規定する酢酸35 mLを水に溶かして100 mLとする。)10 mL
を加えて約70 ℃に温める。この溶液を激しくかき混ぜながら,約70 ℃に温めた塩化バリウム溶液
(JIS K 8155で規定する塩化バリウム二水和物10 gを水に溶かして100 mLとする。)100 mLを1滴
ずつ滴加してクロム酸バリウムを沈殿させ,放置する。上澄み液を捨て,温水約500 mLずつで4回
デカンテーションする。沈殿を遠沈管に移し,遠心分離によって冷水で2〜3回洗浄する。この沈殿を
ガラスろ過器に移して吸引ろ過し,105〜110 ℃で約1時間加熱し,デシケータ中で放冷した後,めの
う乳鉢ですり潰す。
c) カルシウムを含むアンモニア水 JIS K 8122で規定する塩化カルシウム二水和物1.85 gを,アンモニ
ア水(3+4)500 mLに溶かし,ポリエチレン瓶に入れ,空気中の二酸化炭素が入らないような方法で
保存する。図B.4のように保存すると便利である。
d) エタノール(95) JIS K 8102で規定するもの。
e) ジフェニルカルバジド溶液 JIS K 8488で規定する1,5-ジフェニルカルボノヒドラジド(ジフェニル
カルバジド)1 gをエタノール(95)100 mLに溶かす。この溶液は使用時に調製する。
f)
硫酸イオン標準液(SO42−:1 000 mg/L) 17.2.1 g) による。
g) 硫酸イオン標準液(SO42−:10 mg/L) 硫酸イオン標準液(SO42−:1 000 mg/L)1 mLを全量フラス
コ100 mLにとり,水を標線まで加える。
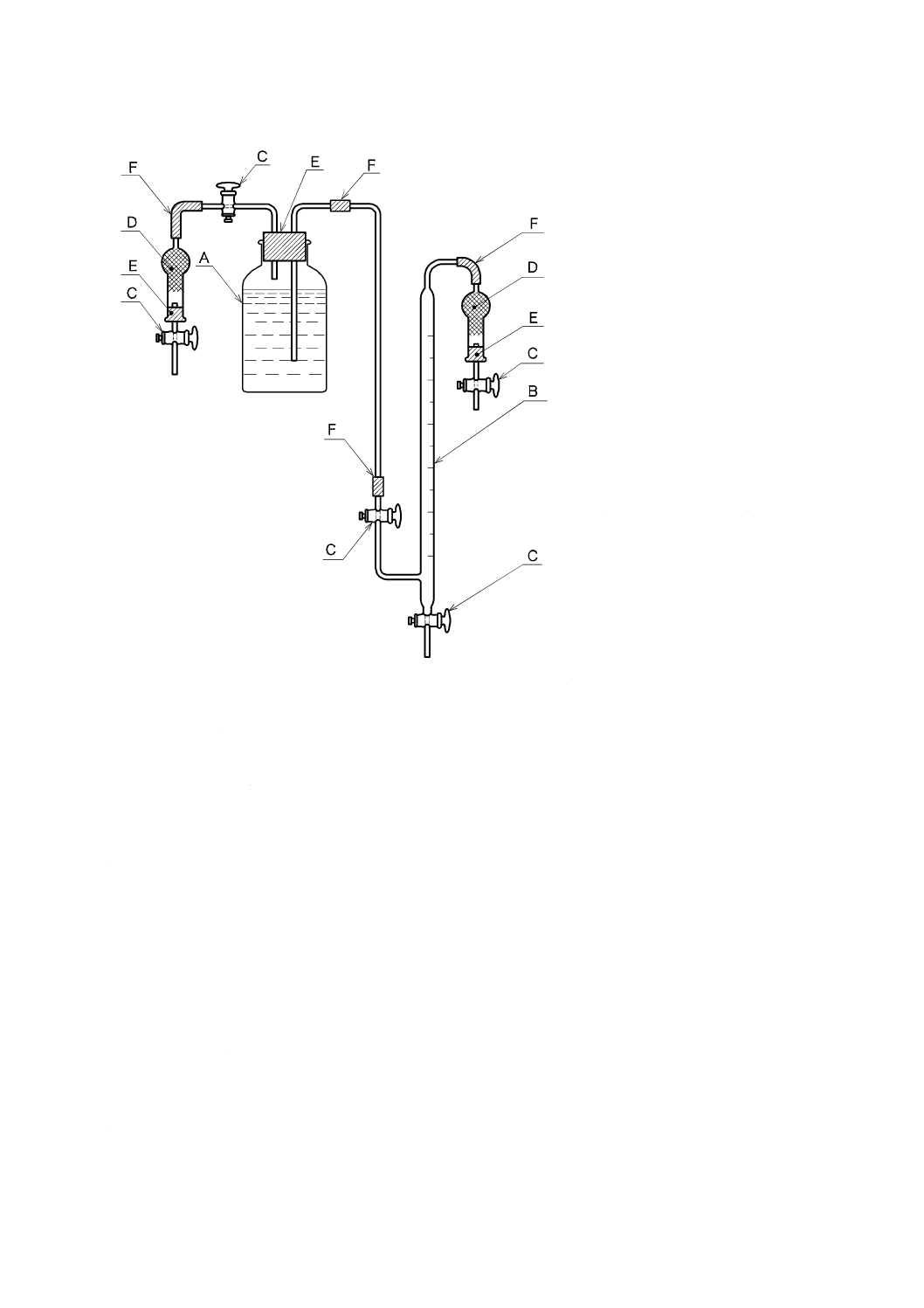
176
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A: ポリエチレン瓶500 mL
B: ビュレット(枝付き)5 mL
C: 二方コック
D: 二酸化炭素吸収管
(ソーダ石灰を詰める。)
E: ゴム栓
F: ゴム管
図B.4−カルシウムを含むアンモニア水の保存の一例
B.8.2.2 器具及び装置
器具及び装置は,次による。
a) ガラスろ過器 ブッフナー漏斗形 3G4
b) 遠心分離器
c) 遠沈管 共栓付き20〜30 mL
d) 光度計 分光光度計又は光電光度計
B.8.2.3 操作
操作は,次による。
a) 試料10 mLを遠沈管にとり,20〜30 ℃に保つ。硫酸イオン濃度が5 mg/L以上の場合には,試料の適
量をとり,水を加えて10 mLとする。これに20〜30 ℃に保ったクロム酸バリウムの酸懸濁液(A)4
mLを加えて振り混ぜ,2〜3分間放置する。
反応時の液温が20〜30 ℃の範囲で同一の検量線が得られる。
b) カルシウムを含むアンモニア水の上澄み液1 mLを図B.4のビュレット又はピペットで静かに加えて
混ぜ,さらにエタノール(95)10 mLを加えて1分間振り混ぜた後,約10分間放置する。
c) これを遠心分離した後,上澄み液をガラスろ過器に移し,弱く吸引ろ過する。
d) ろ液にジフェニルカルバジド溶液2 mLと塩酸(1+1)1.4 mLを加えて振り混ぜ,約5分間放置して
発色させる。
177
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
e) 別に,空試験として水10 mLをとり,試料と同時にa)〜d) の操作を行う。
f)
d) で得た溶液の一部を吸収セルに移し,e) の溶液を対照液として波長540 nm付近の吸光度を測定す
る。
g) あらかじめ,次によって作成した硫酸イオンの検量線から,硫酸イオンの濃度(SO42−:mg/L)を算
出する。a) で試料を適量とり,水で10 mLとした場合は,希釈率を考慮して濃度を算出する。
1) 硫酸イオン標準液(SO42−:10 mg/L)0.2〜5 mLを遠沈管に段階的にとり,水で10 mLとした後,
a)〜f) の操作を行って硫酸イオン濃度(SO42−: mg/L)と吸光度との関係線を作成する。
B.8.3 クロム酸バリウム吸光光度法
試料にクロム酸バリウムの酸懸濁液を加えて硫酸バリウムを沈殿させ,次に,カルシウムイオンを含む
アンモニア水とエタノールとを加え,過剰のクロム酸バリウムを沈殿させ,遠心分離する。硫酸イオンと
置換して生じたクロム酸イオンの黄色の吸光度を測定して硫酸イオンを定量する。
定量範囲:SO42− 5〜50 mg/L,繰返し精度:5〜10 %
B.8.3.1 試薬
試薬は,次による。
a) クロム酸バリウムの酸懸濁液(B) クロム酸バリウム2.5 gを,酢酸(1+15)100 mLと塩酸(1+
500)100 mLとの混合溶液200 mLに加え,よく振り混ぜて懸濁液を作り,ポリエチレン瓶に保存す
る。クロム酸バリウムは,B.8.2.1 b) で調製したものを使用する。
b) カルシウムを含むアンモニア水 B.8.2.1 c) による。
c) エタノール(95) JIS K 8102で規定するもの。
d) 硫酸イオン標準液(SO42−:100 mg /L) 17.2.1 g) の硫酸イオン標準液(SO42−:1 000 mg /L)20 mL
を全量フラスコ200 mLにとり,水を標線まで加える。
e) メチルレッド-ブロモクレゾールグリーン混合液 JIS K 8896で規定するメチルレッド0.02 gとJIS K
8840で規定するブロムクレゾールグリーン0.1 gとをJIS K 8102で規定するエタノール(95)100 mL
に溶かす。
B.8.3.2 器具及び装置
器具及び装置は,次による。
a) 遠心分離器
b) 遠沈管 B.8.2.2 c) による。
c) 光度計 分光光度計又は光電光度計
B.8.3.3 操作
操作は,次による。
a) 試料10 mLを遠沈管にとり,20〜30 ℃に保つ。硫酸イオン濃度が50 mg/L以上の場合には,試料の
適量をとり,水を加えて10 mLとする。これに20〜30 ℃に保ったクロム酸バリウムの酸懸濁液(B)
4 mLを加えて振り混ぜ,2〜3分間放置する。
反応時の液温が20〜30 ℃の範囲で同一の検量線が得られる。
b) カルシウムを含むアンモニア水の上澄み液1 mLを図B.4のビュレット又はピペットで静かに加えて
混ぜ,さらにエタノール(95)10 mLを加えて1分間振り混ぜた後,約10分間放置する。
c) これを遠心分離して,その上澄み液を吸収セルにとり,波長370 nm付近の吸光度を測定する。
d) 空試験として水10 mLをとり,a)〜c) の操作を行って吸光度を測定し,試料について得た吸光度を補
正する。
178
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
e) あらかじめ,次によって作成した硫酸イオンの検量線から,硫酸イオンの濃度(SO42−:mg/L)を算
出する。a) で試料を適量とり,水で10 mLとした場合は,希釈率を考慮して濃度を算出する。
1) 硫酸イオン標準液(SO42−:100 mg/L)0.5〜5 mLを遠沈管に段階的にとり,水で10 mLとした後,
a)〜d) の操作を行って硫酸イオン濃度(SO42−:mg/L)と吸光度との関係線を作成する。
B.8.3.4 留意事項
a) 硝酸,炭酸及び炭酸水素の各イオンは,それぞれ50 mg/L以上共存すると妨害する。りん酸,ひ酸,
セレン酸及びバナジン酸の各イオンと鉛イオンは微量でも妨害するので,前処理を行って除去する。
b) 炭酸イオン及び炭酸水素イオンの除去は,塩酸を加えて煮沸して行う[あらかじめ,試料の一部をと
り,メチルレッド-ブロモクレゾールグリーン混合液を指示薬として塩酸で中和して中和に必要な塩酸
(1+100)(JIS K 8180で規定する塩酸を用いて調製する。)の量を求め,試料に同量の塩酸(1+100)
を加える。塩酸が過剰にならないようにする。]。
c) りん酸イオンを含む場合には,試料10 mLに塩化カルシウム溶液(11 g/L)(JIS K 8123で規定する塩
化カルシウムを用いて調製する。)2 mL及び水酸化ナトリウム溶液(10 g/L)(JIS K 8576で規定する
水酸化ナトリウムを用いて調製する。)と炭酸ナトリウム溶液(13 g/L)(JIS K 8625で規定する炭酸
ナトリウムを用いて調製する。)との等容混合液1 mLを加える。約10分間放置後,遠心分離し,そ
の上澄み液の一定量をとり,塩酸(1+100)で中和した後,水溶中で約10分間加熱して二酸化炭素を
除く。冷却後,水で20 mLに薄め,その10 mLをとり,B.8.3.3の操作によって硫酸イオンを定量する。
このりん酸イオンの除去操作を行った場合の検量線は,硫酸イオン標準液について同様に操作して作
成する。
B.9
りん酸イオン(PO43−)
B.9.1 一般事項
適用頻度が少なく,塩化すず(II)が有害物質であるために,この附属書(参考)に記載する。
B.9.2 モリブデン青[塩化すず(II)還元]吸光光度法
りん酸イオンが七モリブデン酸六アンモニウムと反応して生成するヘテロポリ化合物を塩化すず(II)
で還元し,生成したモリブデン青の吸光度を測定してりん酸イオンを定量する。
定量範囲:PO43− 0.1〜3 mg/L,繰返し精度:2〜10 %
B.9.2.1 試薬
試薬は,次による。
a) 水 JIS K 0557で規定するA3の水
b) モリブデン酸アンモニウム溶液 JIS K 8905で規定する七モリブデン酸六アンモニウム四水和物15 g
を水に溶かし,これを硫酸(水600 mL中にJIS K 8951で規定する硫酸182 mLを静かにかき混ぜなが
ら加えて放冷したもの。)中にかき混ぜながら加え,次に,JIS K 8588で規定するアミド硫酸アンモニ
ウム10 gを加えて溶かした後,水を加えて1 Lとする。試料中に硝酸イオン,亜硝酸イオンが共存し
ない場合には,アミド硫酸アンモニウムを添加しなくてもよい(18.2.1参照)。
c) 塩化すず(II)溶液 JIS K 8136で規定する塩化すず(II)二水和物1 gをJIS K 8180で規定する塩酸
5 mLに溶かし(必要ならば加熱する。),水を加えて50 mLとし,JIS K 8580で規定するすず(粒状)
2,3個を加え,褐色瓶に入れて保存する。濁りを生じたものは使用しない。
d) p-ニトロフェノール溶液(1 g/L) 18.2.1.1 e) による。
e) 硫酸(1+50) 水50容をビーカにとり,これを冷却し,かき混ぜながらJIS K 8951で規定する硫酸
179
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1容を徐々に加える。
f)
水酸化ナトリウム(40 g/L) JIS K 8576で規定する水酸化ナトリウム4 gを水に溶かして100 mLと
する。
g) アンモニア水(1+10) JIS K 8085で規定するアンモニア水を用いて調製する。
h) りん酸イオン標準液(PO43−:100 mg/L) 18.2.1.1 g) による。
i)
りん酸イオン標準液(PO43−:10 mg/L) りん酸イオン標準液(PO43−:100 mg/L)10 mLを全量フラ
スコ100 mLにとり,水を標線まで加える。
B.9.2.2 器具及び装置
器具及び装置は,次による。
a) 光度計 分光光度計又は光電光度計
B.9.2.3 操作
操作は,次による。
a) 5.3によってろ過した試料の40 mLを全量フラスコ50 mLにとる。りん酸イオン濃度が3 mg/L以上の
場合は,試料の適量をとり,水を加えて約40 mLとする。
b) 試料が中性付近にないときは,指示薬としてp-ニトロフェノール溶液(1 g/L)1,2滴を加え,硫酸(1
+50)又は水酸化ナトリウム溶液(40 g/L)若しくはアンモニア水(1+10)を用いて僅かに黄色を呈
するまで中和して一定量とする。また,このとき,鉄(III),アルミニウムなどの水酸化物の沈殿が生
じる場合には,沈殿が生じる直前の微酸性とする。
c) モリブデン酸アンモニウム溶液5 mLを加えて振り混ぜた後,塩化すず(II)溶液0.25 mLを加え,水
を標線まで加えて振り混ぜ,約15分間放置する。
d) 溶液の一部を吸収セルに移し,波長700 nm付近の吸光度を測定する。
e) 空試験として水40 mLをとり,c) 及びd) の操作を行って吸光度を求め,試料について得た吸光度を
補正する。
f)
あらかじめ,次によって作成したりん酸イオンの検量線から,試料中のりん酸イオンの濃度(PO43−:
mg/L)を算出する。a) で試料の適量をとり,水で希釈した場合は,希釈率を考慮して濃度を算出する。
1) りん酸イオン標準液(PO43−:10 mg/L)0.5〜15 mLを全量フラスコ50 mLに段階的にとり,c)〜e) の
操作を行って吸光度を測定し,りん酸イオン濃度(PO43−:mg/L)と吸光度との関係線を作成する。
B.9.2.4 留意事項
a) 塩化物イオン,よう化物イオン,臭化物イオンが共存すると,発色が幾分弱められる。それぞれCl−:
75 mg,I−:6 mg,Br−:25 mgで吸光度の減少は約5 %である。このため,多量のハロゲン化物イオ
ンが共存する場合には,検量線作成時に試料と同量のハロゲン化物イオンを加えるか又はハロゲン化
物のほとんど影響のない18.2.1の方法を用いるとよい。
b) 硫酸イオンが多量に共存すると発色の強さは幾分増加する。モリブデン酸アンモニウム溶液に含まれ
る以外の硫酸イオン500 mgのとき,吸光度は約3 %,1 gのとき約5 %増加する。したがって,多量
の硫酸イオンが含まれる場合は,a) のハロゲン化物イオンの場合と同様に,試料と同量の硫酸イオン
を共存させて検量線を作成するか又は18.2.1の方法を用いるとよい。
c) 亜硫酸イオン150 mgが共存すると約+5 %の正の誤差を生じる。
d) 鉄(III)2 mgが共存するとモリブデン青は,試薬添加後,約15分間で退色し始める。この妨害を防
ぐには,アンモニア水(1+50)又は硝酸(1+25)を用いて試料のpHを約2[水酸化鉄(III)の沈殿
が生成する直前,水酸化鉄(III)が生じた場合は,硝酸(1+25)を滴加して沈殿を溶かす。この場合,
180
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
硝酸(1+25)の過剰量は1 mLを超えないようにする。]とし,これによう化カリウム-亜硫酸ナトリ
ウム溶液(JIS K 8913で規定するよう化カリウム2 g及びJIS K 8061で規定する亜硫酸ナトリウム10
gを水に溶かして100 mLとする。)1 mLを加える。このとき溶液は強い赤褐色となるが,この色が消
えるまで放置する[5〜10分間で消えるが,鉄(III)の多いときは沸騰水浴中に30〜60秒間浸しても
よい。]。この溶液を5.3によってろ過した後,B.9.2.3 a) 以下の操作を行い,りん酸イオンを定量する。
なお,このよう化カリウム-亜硫酸ナトリウム溶液の添加は,硝酸イオン,亜硝酸イオンの影響も除
去する。
e) シリカは,りん酸イオンの500倍量が共存しても誤差は+5 %である。
f)
ろ過しても濁り又は色がある場合は,B.9.2.3 e) の空試験の際に,塩化すず(II)溶液を加えずに操作
して,これを対照液とする。
B.10
アンモニア(NH3)[アンモニウムイオン(NH4+)]
B.10.1 一般事項
アンモニアの定量には,インドフェノール青吸光光度法,イオン電極法,イオンクロマトグラフ法又は
流れ分析法を適用する。
アンモニアはアンモニウムイオンとして測定し,次の換算式を用いてアンモニアへ換算する。
アンモニア(NH3:mg/L)=アンモニウムイオン(NH4+:mg/L)×0.944 1
この試験は試料採取後,直ちに行う。直ちに試験できない場合には,塩酸を加えてpHを2〜3にして0
〜10 ℃の暗所に保存し,できるだけ早く試験する。
化学分析に共通する一般事項は,JIS K 0050による。流れ分析に関わる一般要求事項は,JIS K 0126に
よる。
B.10.2 前処理(蒸留法)
試料に酸化マグネシウムを加えて弱いアルカリ性とし,蒸留を行い,留出したアンモニアを硫酸(25
mmol/L)に吸収捕集する。この方法は,5.3によってろ過しても濁り又は色が残る場合,若しくはカルシ
ウム,マグネシウム,鉄,銅の各イオンなどが多量に含まれる場合に実施する。
B.10.2.1 試薬
試薬は,次による。
a) 水 JIS K 0557で規定するA3の水(使用前に空試験を行い,使用の適否を確認する。)
b) 硫酸(25 mmol/L) JIS K 8951で規定する硫酸約1.4 mLをあらかじめ水100 mLを入れたビーカに
加えてよくかき混ぜ,水を加えて1 Lとする。
c) 硫酸(1+35) JIS K 8951で規定する硫酸を用いて調製する。
d) 水酸化ナトリウム溶液(40 g/L) JIS K 8576で規定する水酸化ナトリウム4 gを水に溶かして100 mL
とする。
e) 酸化マグネシウム JIS K 8432で規定する酸化マグネシウムを使用前に600 ℃で約30分間加熱し,
デシケータ中で放冷する。
f)
ブロモチモールブルー溶液(1 g/L) JIS K 8842で規定するブロモチモールブルー0.1 gをとり,JIS K
8102で規定するエタノール(95)50 mLに溶かし,水で100 mLとする。
B.10.2.2 器具及び装置
器具及び装置は,次による。
蒸留装置 図B.5に例を示す。ガラス器具類は,使用前に水でよく洗う。
181
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
B.10.2.3 操作
操作は,次による。
a) 試料の適量をとり,中性でない場合には,ブロモチモールブルー溶液(1 g/L)5〜7滴を加え,水酸化
ナトリウム溶液(40 g/L)又は硫酸(1+35)でpHを6.0(黄色)〜7.4(青)に調節する。
試料中に残留塩素が存在するときは,蒸留操作の前に,チオ硫酸ナトリウムの小結晶を加えて除去
しておくとよい。
インドフェノール青吸光光度法で定量する場合にはNH4+として0.2 mg/L以上,イオン電極法の場
合には0.2 mg/L以上,イオンクロマトグラフ法の場合は0.1 mg/L以上を含むようにとる。
b) 蒸留フラスコに移し入れ,酸化マグネシウム0.25 g,沸騰石(粒径2〜3 mm)約10個及び水を加えて
液量を約350 mLとする。
c) 蒸留装置を図B.5のように組み立て,受器のメスシリンダ(有栓形)200 mLに硫酸(25 mmol/L)50 mL
を入れる。
d) 蒸留フラスコを加熱し,留出速度5〜7 mL/minで蒸留を行う。冷却器の管の先端は,常に液面下約15
mmを保つようにする。
e) 約140 mLが留出したら蒸留を止める。
f)
冷却器及び逆流止めを外し,冷却器の内管及び逆流止めの内外を少量の水で洗う。洗液は,受器のメ
スシリンダ(有栓形)200 mLに入れ,水を200 mLの標線まで加える。
B.10.2.4 留意事項
a) 蒸留法として水蒸気蒸留法を用いてもよい。この場合は,図B.5の蒸留フラスコに水蒸気を送るよう
に装置を組み立て,蒸留フラスコを加熱する。沸騰し始めたら,水蒸気を蒸留フラスコに送り,留出
速度3〜5 mL/minで蒸留し,約140 mLが留出したら蒸留を止める。
b) 蒸留法においても,脂肪族アミン,芳香族アミン類なども留出するので,これらの共存は妨害となる。
尿素,アセトアミド,ペプトン,アスパラギンなどの窒素を含む有機化合物は,蒸留するとその一部
が加水分解してアンモニアとなり正の誤差を生じる。その程度は,蒸留時のpHが高くなるほど大き
くなる。
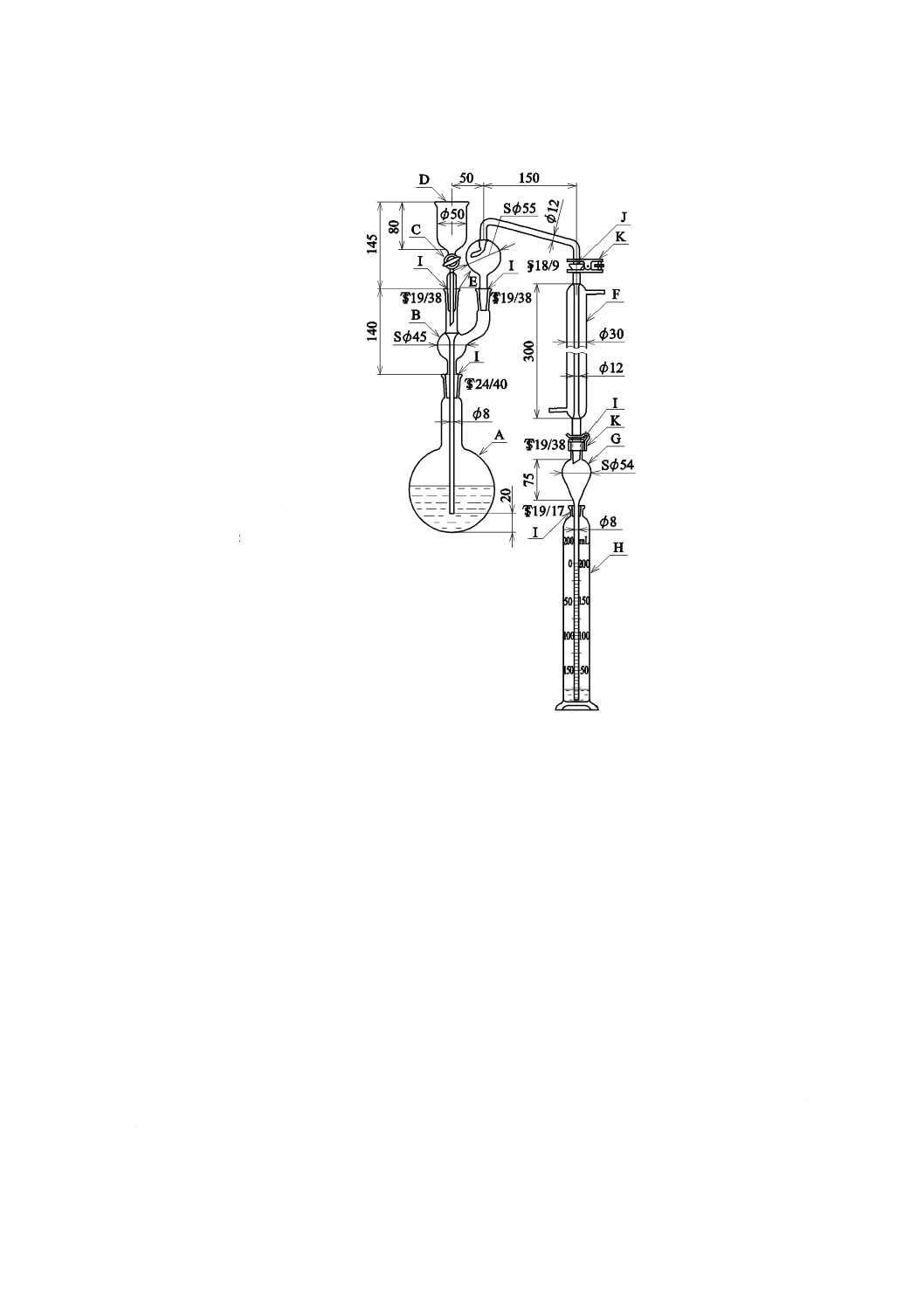
182
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
単位 mm
図B.5−蒸留装置の例
B.10.3 インドフェノール青吸光光度法
アンモニウムイオンが次亜塩素酸イオンの共存の下で,フェノールと反応して生じるインドフェノール
青の吸光度を測定してアンモニウムイオンを定量する。
定量範囲:NH4+:0.2〜4 mg/L,繰返し精度:2〜10 %
なお,微量のアンモニウムイオンを定量する場合には,B.10.3.3 b) の操作でナトリウムフェノキシド溶
液10 mLに続き,ペンタシアノニトロシル鉄(III)酸二ナトリウム溶液[JIS K 8722で規定するペンタシ
アノニトロシル鉄(III)酸ナトリウム二水和物0.15 gを水に溶かして100 mLとする。]1 mLを加えると
よい。
この場合の定量範囲は,NH4+として0.1〜2 mg/Lとなる。検量線は,同一操作で作成する。
B.10.3.1 試薬
試薬は,次による。
a) 水 B.10.2.1 a) による。
b) EDTA溶液 JIS K 8107で規定するエチレンジアミン四酢酸二水素二ナトリウム二水和物5 gを水に
溶かして100 mLとする。
c) 水酸化ナトリウム溶液(200 g/L) JIS K 8576で規定する水酸化ナトリウム20 gを水に溶かして100
A: 蒸留フラスコ500 mL
B: 連結導入管
C: すり合わせコック
D: 注入漏斗
E: トラップ球(ケルダール球)
F: リービッヒ冷却器300 mm
G: 逆流止め(約50 mL)
H: 受器[メスシリンダ(有栓形)200 mL]
I: 共通すり合わせ
J: 共通球面すり合わせ
K: 押さえばね
183
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
mLとする。使用時に調製する。
d) ナトリウムフェノキシド溶液 c) の水酸化ナトリウム溶液(200 g/L)55 mLをビーカにとり,冷水中
で冷却しながら,JIS K 8798で規定するフェノール25 gを少量ずつ加えて溶かす。放冷後,JIS K 8034
で規定するアセトン6 mLを加え,水を加えて200 mLとする。10 ℃以下の暗所に保存し,5日間以
上経過したものは使用しない。
e) 次亜塩素酸ナトリウム溶液(有効塩素10 g/L) 次亜塩素酸ナトリウム溶液(有効塩素7〜12 %)の
有効塩素の濃度を次の標定操作に従って求め,有効塩素が約10 g/Lになるように水で薄める。使用時
に調製する。この溶液は使用時に,次によって標定して用いる。
1) 次亜塩素酸ナトリウム溶液(有効塩素7〜12 %)10 mLを全量フラスコ200 mLにとり,水を標線ま
で加える。この10 mLを共栓三角フラスコ300 mLにとり,水を加えて約100 mLとする。JIS K 8913
で規定するよう化カリウム1〜2 g及び酢酸(1+1)[JIS K 8355で規定する酢酸を用いて調製する。]
6 mLを加えて密栓し,よく振り混ぜて暗所に約5分間放置した後,50 mmol/Lチオ硫酸ナトリウム
溶液で滴定する。溶液の黄色が薄くなったら,指示薬としてでんぷん溶液(10 g/L)約l mLを加え,
生じたよう素でんぷんの青い色が消えるまで滴定する。別に,空試験として水10 mLをとり,同じ
操作を行って滴定値を補正する。次の式によって有効塩素(g/L)を算出する。
773
001
.0
000
1
10
200
×
×
×
×
V
f
a
N=
ここに,
N: 有効塩素(g/L)
a: 滴定に要した50 mmol/Lチオ硫酸ナトリウム溶液(mL)
f: 50 mmol/Lチオ硫酸ナトリウム溶液のファクタ
0.001 773: 50 mmol/Lチオ硫酸ナトリウム溶液1 mLの有効塩素相
当量(g)
V: 次亜塩素酸ナトリウム溶液(有効塩素7〜12 %)(mL)
f)
アンモニウムイオン標準液(NH4+:1 000 mg/L) 21.8.1 e) による。
g) アンモニウムイオン標準液(NH4+:10 mg/L) アンモニウムイオン標準液(NH4+:1 000 mg/L)10 mL
を全量フラスコ1 000 mLにとり,水を標線まで加える。使用時に調製する。
h) 塩酸(1+1) JIS K 8180で規定する塩酸を用いて調製する。
B.10.3.2 器具及び装置
器具及び装置は,次による。
a) メスシリンダ(有栓形) 15.2.2 b) による。使用前にB.10.2.1 a) の水でよく洗う。
b) 光度計 分光光度計又は光電光度計
B.10.3.3 操作
操作は,次による。
a) 5.3によってろ過した試料又はB.10.2で処理した試料25 mLを全量フラスコ50 mLにとる。アンモニ
ウムイオン濃度が4 mg/L以上の場合は,試料の適量をとり,水を加えて25 mLとする。
酸消費量の高い試料には,塩酸(1+1)を用いて中和(pH約7まで)する。発色の強さは,pH 11.5
〜12.5のとき最高となるのでアルカリ性の強い試料は,あらかじめ中和しておく。
b) EDTA溶液1 mLとナトリウムフェノキシド溶液10 mLとを加えて振り混ぜる。EDTA溶液を添加する
とインドフェノール青の発色がやや弱くなるので,検量線作成時にも同一量のEDTA溶液を添加する。
c) 次亜塩素酸ナトリウム溶液(有効塩素10 g/L)5 mLを加え,水を50 mLの標線まで加え,栓をして振
り混ぜる。
184
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
d) 液温を20〜25 ℃に保って約30分間放置する。液温が20〜25 ℃のとき約30分間で発色は最高となり,
その後,約30分間は安定である。
e) この溶液の一部を吸収セルに移し,波長630 nm付近の吸光度を測定する。
f)
空試験として水25 mLをとり,b)〜e) の操作を行って吸光度を測定し,試料について得た吸光度を補
正する。
g) あらかじめ,次によって作成したアンモニウムイオンの検量線から,試料中のアンモニウムイオンの
濃度(NH4+:mg/L)を算出する。a) で試料の適量をとり,水で希釈した場合は,希釈率を考慮して
濃度を算出する。
1) アンモニウムイオン標準液(NH4+:10 mg/L)0.5〜10 mLを段階的に全量フラスコ50 mLにとり,
a)〜f) の操作を行って吸光度を測定し,アンモニウムイオンの濃度(NH4+:mg/L)と吸光度との
関係線を作成する。
B.10.3.4 留意事項
a) 鉄(II)及び銅(II)は,いずれも0.15 mg/Lまでは妨害しない。また,それぞれ1 mg/L以下であれば,
EDTA溶液の添加で妨害を除くことができる。
b) 脂肪族アミン類は妨害しないが,芳香族アミン類の一部は次亜塩素酸塩によって酸化され着色物質を
生じるので妨害する。また,p-アミノフェノールのような物質は,アルカリ性溶液中でフェノールと
反応してインドフェノール青を生じるので妨害する。p-ヒドロキノンは妨害しない。ヒドロキシルア
ミンも妨害するが,JIS K 8230で規定する過酸化水素(30 %)を定量的に加えて酸化すれば,妨害を
除くことができる。
B.10.4
イオン電極法
試料に水酸化ナトリウム溶液を加え,pHを11〜13にしてアンモニウムイオンをアンモニアに変え,ア
ンモニア電極を指示電極として電位を測定し定量する。
定量範囲:NH4+:0.1〜100 mg/L,繰返し精度:5〜20 %
B.10.4.1 試薬
試薬は,次による。
a) 水 B.10.2.1 a) による。
b) 水酸化ナトリウム溶液(100 g/L) JIS K 8576で規定する水酸化ナトリウム10 gを水に溶かして100
mLとする。
c) アンモニウムイオン標準液(NH4+:100 mg/L) 21.8.1 e) のアンモニウムイオン標準液(NH4+:1 000
mg/L)20 mLを全量フラスコ200 mLにとり,水を標線まで加える。
d) アンモニウムイオン標準液(NH4+:10 mg/L) アンモニウムイオン標準液(NH4+:100 mg/L)20 mL
を全量フラスコ200 mLにとり,水を標線まで加える。
e) アンモニウムイオン標準液(NH4+:1 mg/L) アンモニウムイオン標準液(NH4+:10 mg/L)20 mL
を全量フラスコ200 mLにとり,水を標線まで加える。使用時に調製する。
f)
アンモニウムイオン標準液(NH4+:0.1 mg/L) アンモニウムイオン標準液(NH4+:1 mg/L)20 mL
を全量フラスコ200 mLにとり,水を標線まで加える。使用時に調製する。
B.10.4.2 器具及び装置
器具及び装置は,次による。
a) 電位差計 15.5.2 a) による。
b) イオン濃度計
185
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
c) 指示電極 アンモニア電極(隔膜形)
d) 参照電極 塩化カリウムを充塡した銀-塩化銀電極を用いる。
e) 測定容器 15.5.2 e) による。アンモニアは揮散しやすいため,できるだけ口の狭い容器を用いる。必
要に応じて密閉できる容器を用いるとよい。
f)
恒温槽 15.5.2 f) による。
g) マグネチックスターラ 15.5.2 g) による。
B.10.4.3 操作
B.10.4.3.1 イオン電極を用いる場合
操作は,次による。
a) 5.3によってろ過した試料又はB.10.2で処理した試料100 mLを測定容器200 mLにとり,水酸化ナト
リウム溶液(100 g/L)1 mLを加える。水酸化ナトリウム溶液(100 g/L)は電位の測定の直前に加え
る。
アンモニウムイオンを100 mg/L以上含む場合は,試料量を減らし,水を加えて100 mLとする。
b) 恒温槽から水を送り,液温を25±0.5 ℃に調節する。
c) 指示電極と参照電極とを浸し,マグネチックスターラで泡が電極に触れない程度に強くかき混ぜる。
マグネチックスターラを長時間使用する場合には,発熱して液温に変化を与えることがあるので,液
温の変化に注意する。また,かき混ぜが強すぎると,泡が膜を覆い誤差を生じるので注意する。
隔膜形電極は,内部ガラス電極のガラス膜面に隔膜を強く押し付けると隔膜にきずがつくので注意
する。また,ガラス膜面と隔膜が離れ過ぎると,応答時間が長くなる。電極の隔膜が汚れると,電位
が不安定になり,応答時間も長くなる。
d) 液温を確認し,電位差計で電位を測定する。
e) あらかじめ,次によって作成したアンモニウムイオンの検量線から,試料中のアンモニウムイオンの
濃度(NH4+:mg/L)を算出する。
1) アンモニウムイオン標準液(NH4+:0.1 mg/L)100 mLを測定容器200 mLにとり,水酸化ナトリウ
ム溶液(100 g/L)1 mLを加える。水酸化ナトリウム溶液(100 g/L)は電位の測定の直前に加える。
2) b)〜d) の操作を行い,液温を確認し,電位差計で電位を測定する。アンモニア電極の応答時間は,
液温10〜30 ℃の場合アンモニウムイオン標準液(NH4+:0.1 mg/L)で3〜5分間,アンモニウムイ
オン標準液(NH4+:1 mg/L以上)では2〜3分間である。
3) アンモニウムイオン標準液(NH4+:1 mg/L)100 mL,アンモニウムイオン標準液(NH4+:10 mg/L)
100 mL及びアンモニウムイオン標準液(NH4+:100 mg/L)100 mLをそれぞれ三角フラスコ200 mL
にとり,水酸化ナトリウム溶液(100 g/L)1 mLを加える。
4) b)〜d) の操作を行い,それぞれのアンモニウムイオン標準液の電位を測定する。電位の測定は,低
濃度から順に行う。高濃度から低濃度へ移ると,応答速度は遅くなるので,水で洗ってアンモニア
を除き,次に,B.10.4.1 b) の水酸化ナトリウム溶液(100 g/L)1 mLを加えたアンモニウムイオン
標準液(NH4+:0.1 mg/L)に浸し,指示値が安定してから測定する。
なお,水酸化ナトリウムを加えた標準液は,5〜10分間経過するとアンモニアを消失しているの
で,次の校正には新しい標準液を用いる。
5) 横軸にアンモニウムイオンの濃度(NH4+:mg/L)の対数を,縦軸に電位をとり,アンモニウムイ
オンの濃度(NH4+:mg/L)と電位との関係線を作成する。電位は,ネルンスト式に従って,アン
モニウムイオンの濃度が10倍の変化当たり約60 mV変化する。アンモニウムイオンの濃度NH4+:
186
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
0.1 mg/L付近からNH4+:100 mg/Lまで検量線は直線になる。
B.10.4.3.2 イオン濃度計を用いる場合
操作は,次による。
a) アンモニウムイオン標準液(NH4+:0.1 mg/L)とアンモニウムイオン標準液(NH4+:10 mg/L)を別々
の測定容器にとり,水酸化ナトリウム溶液(100 g/L)1 mLを加える。水酸化ナトリウム溶液(100 g/L)
は測定の直前に加える。
b) B.10.4.3.1のb) 及びc) の操作を行い,イオン濃度計の指示値をNH4+:0.1 mg/LとNH4+:10 mg/Lと
になるように調節する。
c) さらにアンモニウムイオン標準液(NH4+:1 mg/L)とアンモニウムイオン標準液(NH4+:100 mg/L)
とを用いてイオン濃度計の指示値を確認する。
d) B.10.4.3.1のa)〜d) の操作を行い,イオン濃度計の指示値を読み,試料のアンモニウムイオン濃度
(NH4 +:mg/L)を求める。
B.10.4.4 留意事項
a) 試料中のアンモニウムイオンの濃度がNH4+:0.1 mg/Lの場合,ヒドラジニウムイオンはN2H5+:1 mg/L
以下では影響しないが,10 mg/L及び100 mg/L共存すれば,それぞれ+35 %,+100 %の誤差を生じ
る。
b) 試料中のアンモニウムイオンの濃度がNH4+:1 mg/L以上の場合には,ヒドラジニウムイオン100 mg/L
が共存しても影響しない。
c) 試料中のアンモニウムイオンの濃度がNH4+:0.1 mg/Lの場合,モルホリン[テトラヒドロ-1,4-オキサ
ジン(C4H8ONH)]は10 mg/Lまで共存しても影響しないが,100 mg/L共存すると,100 %の誤差を生
じる。
d) 試料中のアンモニウムイオンの濃度が1 mg/L以上の場合には,モルホリンが100 mg/L共存しても影
響しない。
e) アンモニア電極は,アンモニウムイオンの濃度が50 mg/Lを超える試料の測定に連続的に使用すると,
満足な応答をしない。このような水に対しては,測定試料をこの濃度未満に薄めるのがよい。
f)
アミン類は正の妨害を与える。この他の妨害物質として表B.7に示すヒドラジン,シクロヘキシルア
ミン,モルホリン,オクタデシルアミン,メタノールアミン,尿素などがある。
g) 界面活性剤及びある種の有機溶媒は,アンモニア電極の膜の寿命を短くする。このため電極の手入れ
を必要とする頻度が増加する。この影響はかなり重大で,これらの妨害物質の濃度が高い試料では,
アンモニア電極の劣化を早めることになる。
表B.7−妨害物質の例
妨害物質
妨害物質濃度
mg/L
アンモニウムイオンの濃度ρN=
1 mg/Lにおける見かけの増加
mg/L
ヒドラジン
4
0.06
シクロヘキシルアミン
1
0.03
モルホリン
10
0.03
オクタデシルアミン
0.4
0.14
メタノールアミン
3.4
0.15(ρN=0.5 mg/L)
尿素
11
0.01
187
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
B.10.5 イオンクロマトグラフ法
試料中のアンモニウムイオンをイオンクロマトグラフ法によって定量する。この方法を適用する場合に
は,一般に試料採取後,直ちに試験する。
定量範囲:NH4+:0.1〜30 mg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
B.10.5.1 試薬
試薬は,21.8.1による。ただし,標準液は,次による。
a) アンモニウムイオン標準液(NH4+:100 mg/L) B.10.4.1 c) による。
B.10.5.2 器具及び装置
器具及び装置は,21.8.2による。
B.10.5.3 準備操作
準備操作は,21.8.3による。
B.10.5.4 操作
操作は,次による。
a) イオンクロマトグラフを定量できる状態にし,分離カラムに溶離液を一定の流量(例えば,1〜2
mL/min)で流しておく。サプレッサを必要とする装置では再生液を一定の流量で流しておく。
b) B.10.5.3の準備操作を行った試料の一定量(例えば,50〜200 µLの一定量)をイオンクロマトグラフ
に注入し,クロマトグラムを記録する。
c) クロマトグラム上のアンモニウムイオンに相当するピークについて,指示値(ピーク高さ又はピーク
面積)を読み取る。
d) 試料を薄めた場合には,空試験として試料と同量の水について,a)〜c) の操作を行って試料について
得た結果を補正する。
e) あらかじめ,次によって作成したアンモニウムイオンの検量線から,試料中のアンモニウムイオンの
濃度(NH4+:mg/L)を算出する。
1) アンモニウムイオン標準液(NH4+:100 mg/L)又は陽イオン混合標準液[(NH4+:100 mg,Na:100
mg,K:100 mg,Ca:200 mg,Mg:200 mg)/L]0.1〜30 mLを段階的に全量フラスコ100 mLにと
り,水を標線まで加える。この溶液についてa)〜c) の操作を行ってそれぞれのアンモニウムイオン
に相当する指示値(ピーク高さ又はピーク面積)を読み取る。別に,空試験としてこの操作に用い
た水についてa)〜c) の操作を行ってそれぞれのアンモニウムイオンに相当する指示値を補正した
後,アンモニウムイオン(NH4+)の濃度と指示値との関係線を作成する。検量線の作成は測定時に
行う。
B.10.5.5 留意事項
a) アンモニウムイオンの濃度がNH4+:1 mg/Lのときナトリウム及びカリウムはいずれも100 mg/L以下
ならば妨害しない。
b) カルボン酸形の陽イオン交換カラムと溶離液としてメタンスルホン酸,[2,6-ピリジンカルボン酸
-L(+)-酒石酸]溶液などを用いると,カルシウム,マグネシウムなど2価の陽イオンの溶離及び定量
が可能になる。
B.10.6
流れ分析法
試料中のアンモニウムイオンを,B.10.3と同様な原理で発色させる流れ分析法によって定量する。試料
を,B.10.2の操作でアンモニウムイオンを共存物から分離した後,適用する。ただし,妨害物質を含まな
い試料は,蒸留操作を省略してもよい。この場合は懸濁物の多い試料には適用できない。
188
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
定量範囲:NH4+:0.06〜13 mg/L(アンモニア体窒素として0.05〜10 mg/L),繰返し精度:10 %以下(装
置及び測定条件によって異なる。)
B.10.6.1 フェノールによるインドフェノール青発色FIA法
細管中を連続して流れているキャリヤ液中に試料を注入し,フェノール・ペンタシアノニトロシル鉄(III)
酸ナトリウム溶液及び次亜塩素酸ナトリウム溶液の流れを順次合流させ,反応させる。反応によって生成
する青い色の化合物の660 nm付近の吸光度を測定し,アンモニウムイオンを定量する。
B.10.6.1.1 試薬
試薬は,次による。調製した溶液は,必要に応じて脱気を行う。
a) キャリヤ液 JIS K 0557で規定するA3又はA4の水を用いる(使用前に空試験を行い,使用の適否を
確認する。)。
b) フェノール・ペンタシアノニトロシル鉄(III)酸ナトリウム溶液 JIS K 8798で規定するフェノール
20 gとJIS K 8722で規定するペンタシアノニトロシル鉄(III)酸ナトリウム二水和物0.1 gとを水約
900 mLに溶かし,水を加えて1 000 mLとする。
なお,フェノールの代わりにサリチル酸ナトリウムを用いることができる。サリチル酸ナトリウム・
ペンタシアノニトロシル鉄(III)酸ナトリウム溶液は,水約800 mLにJIS K 8397で規定するサリチ
ル酸ナトリウム100 g,JIS K 8536で規定する(+)-酒石酸ナトリウムカリウム四水和物19 g及びJIS K
8722で規定するペンタシアノニトロシル鉄(III)酸ナトリウム二水和物0.75 gを溶かし,水を加えて
1 000 mLとする。
c) 水酸化ナトリウム溶液(20 g/L) JIS K 8576で規定する水酸化ナトリウム20 gを水に溶かして1 000
mLとする。
d) 次亜塩素酸ナトリウム溶液 水約900 mLに次亜塩素酸ナトリウム溶液(有効塩素濃度5 %)10 mL相
当量と水酸化ナトリウム15 gとを溶かし,水を加えて1 000 mLとする。この溶液は使用時に調製す
る。次亜塩素酸ナトリウム溶液の有効塩素は,B.10.3.1 e) によって求め,次亜塩素酸ナトリウム溶液
(有効塩素濃度5 %)10 mL相当量を求める。
なお,フェノール・ペンタシアノニトロシル鉄(III)酸ナトリウム溶液の代わりにサリチル酸・ペ
ンタシアノニトロシル鉄(III)酸ナトリウム溶液を用いた場合は,次亜塩素酸ナトリウム溶液(有効
塩素濃度5 %)100 mL相当量と水酸化ナトリウム溶液(20 g/L)900 mLとを混合したものを用いる。
次亜塩素酸ナトリウム溶液の有効塩素は,B.10.3.1 e) によって求め,次亜塩素酸ナトリウム溶液(有
効塩素濃度5 %)100 mL相当量を求める。
e) 硫酸(1 mol/L) 水900 mLをビーカにとり,これをかき混ぜながらJIS K 8951で規定する硫酸55 mL
を徐々に加え,放冷後,水で1 000 mLにする。
f)
アンモニウムイオン標準液(NH4+:1 000 mg/L) 21.8.1 e) による。
g) アンモニウムイオン標準液(NH4+:100 mg/L) アンモニウムイオン標準液(NH4+:1 000 mg/L)10
mLを正確にビーカにとり,水約80 mLを加え,硫酸(1 mol/L)を添加して,pH 2に調節した後,全
量フラスコ100 mLに水とともに移し入れ,水を標線まで加える。この溶液は,冷暗所で保存する。
h) アンモニウムイオン標準液(NH4+:10 mg/L) アンモニウムイオン標準液(NH4+:1 000 mg/L)1 mL
又はアンモニウムイオン標準液(NH4+:100 mg/L)10 mLを正確にビーカにとり,水約80 mLを加え,
硫酸(1 mol/L)を添加して,pH 2に調節した後,全量フラスコ100 mLに水とともに移し入れ,水を
標線まで加える。この溶液は,冷暗所で保存する。
B.10.6.1.2 器具及び装置
189
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
装置の基本構成は,次による(図B.6参照)。
a) 送液部 15.7.1.2 a) による。
b) 試料導入部 15.7.1.2 b) による。
c) 反応部 15.7.1.2 c) 及び一定の温度に加熱保持できる恒温槽から構成する。
d) 検出部 波長660 nm付近での測定が可能な吸光光度検出器を用いる。
e) 記録部 15.7.1.2 e) による。
f)
恒温槽 80 ℃に加熱できるもの。
C: キャリヤ液
R1: フェノール・ペンタシアノニトロシル鉄(III)酸ナトリウム溶液
R2: 次亜塩素酸ナトリウム溶液
S: 試料
1: ポンプ
2: 試料導入器(200 μL)
3: 反応コイル(内径0.5 mm,長さ1 m)
4: 恒温槽(80 ℃)
5: 反応コイル(内径0.5 mm,長さ10 m)
6: 検出器(吸収セル 光路長1 cm,波長660 nm)
7: 廃液
図B.6−フェノールによるインドフェノール青発色FIA法のシステム例
B.10.6.1.3 準備操作
準備操作は,15.7.1.3による。ただし,感度の調節はアンモニウムイオンによる。
B.10.6.1.4 操作
操作は,15.7.1.4による。ただし,検量線は,B.10.6.1.1 h) を水で希釈して調製した検量線用アンモニウ
ムイオン標準液を用いて作成し,試料中のアンモニウムイオンの濃度の算出には,これを用いる。
B.10.6.1.5 留意事項
留意事項は,15.7.1.5による。
B.10.6.2 フェノールによるインドフェノール青発色CFA法
細管中を連続して流れている試料に,フェノール溶液,ペンタシアノニトロシル鉄(III)酸ナトリウム
溶液及び次亜塩素酸ナトリウム溶液の流れを細管中で順次合流させ,反応させる。反応によって生成する
青い色の化合物の630 nm付近の吸光度を測定し,アンモニウムイオンを定量する。
B.10.6.2.1 試薬
試薬は,次による。調製した溶液は,必要に応じて脱気を行う。
190
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) ドデシル硫酸ナトリウム溶液(150 g/L) ドデシル硫酸ナトリウム15 gを水に溶かして100 mLとす
る。
b) 水酸化ナトリウム溶液(1 mol/L) JIS K 8576で規定する水酸化ナトリウム40 gを水に溶かして1 000
mLとする。
c) 塩酸(1 mol/L) JIS K 8180で規定する塩酸10 mLをとり,水を加えて120 mLとする。
d) CyDTA溶液 JIS K 8576で規定する水酸化ナトリウム8 g,JIS K 8625で規定する炭酸ナトリウム5 g
及びCyDTA 22 gを水約800 mLに溶かし,水酸化ナトリウム溶液(1 mol/L)又は塩酸(1 mol/L)で
pH 10±0.1に調節し,水で1 000 mLとする。
e) ペンタシアノニトロシル鉄(III)酸ナトリウム溶液 JIS K 8722で規定するペンタシアノニトロシル
鉄(III)酸ナトリウム二水和物0.15 gを水に溶かし250 mLとする。
f)
CyDTA混合溶液 CyDTA溶液160 mL,ペンタシアノニトロシル鉄(III)酸ナトリウム溶液40 mL及
びドデシル硫酸ナトリウム溶液(150 g/L)2 mLを混合する。
g) フェノール溶液 JIS K 8798で規定するフェノール47.5 g及びJIS K 8576で規定する水酸化ナトリウ
ム19 gを水に溶かして,500 mLとする。
h) 次亜塩素酸ナトリウム溶液 次亜塩素酸ナトリウム溶液(有効塩素濃度約5 %)30 mLに水を加えて
100 mLとする。この溶液は使用時に調製する。次亜塩素酸ナトリウム溶液の有効塩素は,B.10.3.1 e) に
よって求め,次亜塩素酸ナトリウム溶液(有効塩素濃度5 %)30 mL相当量を求める。
i)
硫酸(1 mol/L) B.10.6.1.1 e) による。
j)
アンモニウムイオン標準液(NH4+:1 000 mg/L) 21.8.1 e) による。
k) アンモニウムイオン標準液(NH4+:100 mg/L) B.10.6.1.1 g) による。
l)
アンモニウムイオン標準液(NH4+:10 mg/L) B.10.6.1.1 h) による。
B.10.6.2.2 器具及び装置
装置の基本構成は,次による(図B.7参照)。
a) 送液部 15.7.1.2 a) による。
b) 試料導入部 15.7.2.2 b) による。
c) 反応部 15.7.2.2 c) 及び一定の温度に加熱保持できる恒温槽から構成する。
d) 検出部 波長630 nm付近での測定が可能な吸光光度検出器を用いる。
e) 記録部 15.7.1.2 e) による。
f)
恒温槽 45 ℃に加熱できるもの。
191
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
R1: CyDTA混合溶液
R2: フェノール溶液
R3: 次亜塩素酸ナトリウム溶液
S: 試料又は水
1: ポンプ
2: セグメントガス(空気)
3: 反応コイル(内径1.0 mm,長さ27 cm)
4: 恒温槽(45 ℃)
5: 反応コイル(内径1 mm,長さ3 m)
6: 反応コイル(内径1 mm,長さ54 cm)
7: 検出器(波長 630 nm付近)
8: 廃液
図B.7−フェノールによるインドフェノール青発色CFA法のシステム例
B.10.6.2.3 準備操作
準備操作は,15.7.2.3による。ただし,感度の調節はアンモニウムイオンによる。
B.10.6.2.4 操作
操作は,15.7.2.4による。ただし,検量線は,B.10.6.2.1 l) を水で希釈して調製した検量線用アンモニウ
ムイオン標準液を用いて作成し,試料中のアンモニウムイオンの濃度の算出には,これを用いる。
B.10.6.2.5 留意事項
留意事項は,15.7.2.5による。
B.11
ニッケル(Ni)
B.11.1
一般事項
ニッケルの定量には,ジメチルグリオキシム吸光光度法,フレーム原子吸光法又はICP発光分光分析法
を適用する。
B.11.2
ジメチルグリオキシム吸光光度法
試料にくえん酸塩を加え,アンモニア水で微アルカリ性とした後,2,3-ブタンジオンジオキシム(ジメ
チルグリオキシム)を加えて生成したニッケル錯体をクロロホルムで抽出し,これを希塩酸で逆抽出する。
抽出液に臭素及びアンモニア水を加えてニッケルを酸化し,再び2,3-ブタンジオンジオキシムを加えて生
じる赤褐色のニッケル錯体の吸光度を測定してニッケルを定量する。
192
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
定量範囲:Ni:4〜100 μg/L(試料500 mLを使用する場合),繰返し精度:2〜10 %
B.11.2.1 試薬
試薬は,次による。
a) 水 21.2.1 a) による。
b) 塩酸(1+20) JIS K 8180で規定する塩酸を用いて調製する。
c) アンモニア水(1+1)及び(1+5) JIS K 8085で規定するアンモニア水を用いて調製する。
d) 臭素水(飽和) JIS K 8529で規定する臭素3〜4 mLを水100 mLに加えて激しく振り混ぜ,放置後
その上澄み液を用いる。
e) くえん酸水素二アンモニウム溶液(100 g/L) JIS K 8284で規定するくえん酸水素二アンモニウム10
gを水約80 mLに溶かし,アンモニア水(1+1)を滴加してpHを約7に調節した後,水を加えて100
mLとする。
f)
フェノールフタレイン溶液(5 g/L) 9.3.1 a) による。
g) ジメチルグリオキシムのエタノール溶液(10 g/L) JIS K 8498で規定するジメチルグリオキシム1 g
をJIS K 8102で規定するエタノール(95)に溶かして100 mLとする。
h) ジメチルグリオキシムの水酸化ナトリウム溶液(10 g/L) JIS K 8498で規定するジメチルグリオキ
シム1 gを水酸化ナトリウム溶液(10 g/L)(JIS K 8576で規定する水酸化ナトリウムを用いて調製す
る。)に溶かし,水酸化ナトリウム溶液(10 g/L)を加えて100 mLとする。不溶解物があるときはろ
過する。
i)
クロロホルム JIS K 8322で規定するもの。
j)
ニッケル標準液(Ni:100 mg/L) 24.7.1 e) による。
k) ニッケル標準液(Ni:5 mg/L) ニッケル標準液(Ni:100 mg/L)50 mLを全量フラスコ1 000 mLに
とり,硝酸(1+1)[21.6.1 b) による。]20 mLを加え,水を標線まで加える。
B.11.2.2 器具及び装置
器具及び装置は,次による。
a) 分液漏斗
b) 光度計 分光光度計又は光電光度計
B.11.2.3 操作
操作は,次による。
a) 箇条6の操作を行った試料の適量(Niとして4〜100 μg/Lを含む。)を分液漏斗にとり,くえん酸水素
二アンモニウム溶液(100 g/L)5 mL及び指示薬としてフェノールフタレイン溶液(5 g/L)2,3滴を
加え,次に,アンモニア水(1+5)を溶液の色が僅かに赤い色になるまで滴加する。さらに,アンモ
ニア水(1+5)2,3滴及び水を加えて約100 mLとする。
b) ジメチルグリオキシムのエタノール溶液(10 g/L)2 mLとクロロホルム10 mLとを加え,約1分間激
しく振り混ぜて放置した後,クロロホルム層を別の分液漏斗に入れる。水層にクロロホルム5 mLを
加え,約1分間激しく振り混ぜて抽出し,放置後,クロロホルム層をとり,先の分液漏斗に合わせる。
この操作を更に1回繰り返す。
c) クロロホルム層を入れた分液漏斗にアンモニア水(1+50)(JIS K 8085で規定するアンモニア水を用
いて調製する。)10〜20 mLを加え,約30秒間振り混ぜて放置した後,クロロホルム層を別の分液漏
斗に移す。
d) クロロホルム層を入れた分液漏斗に塩酸(1+20)10 mLを加え,約1分間激しく振り混ぜ,ニッケル
193
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
を逆抽出する。放置後,クロロホルム層を別の分液漏斗に入れる。クロロホルム層には,再び塩酸(1
+20)5 mLを加え,逆抽出を繰り返す。クロロホルム層は捨て,水層は先の水層に合わせて全量フラ
スコ25 mLに入れる。
e) 臭素水(飽和)2 mLを加えて振り混ぜ,約1分間放置する。次に,アンモニア水(1+1)を加えて中
和し,更にアンモニア水(1+1)2 mLを過剰に加え,流水で室温以下に冷却する。
f)
ジメチルグリオキシムの水酸化ナトリウム溶液(10 g/L)2 mLを加えて振り混ぜ,ニッケルを発色さ
せた後,水を標線まで加える。
g) この溶液の一部を吸収セルに移し,波長450 nm付近の吸光度を測定する。
h) 空試験として水約50 mLをとり,a)〜g) の操作を行って吸光度を測定し,試料について得た吸光度を
補正する。
i)
あらかじめ,次によって作成したニッケルの検量線から,試料中のニッケルの濃度(μg/L)を算出す
る。
1) 検量線の作成は,ニッケル標準液(Ni:5 mg/L)0.4〜10 mLを全量フラスコ25 mLに段階的にとり,
a)〜h) の操作を行ってニッケル(Ni)の濃度と吸光度との関係線を作成する。
B.11.2.4 留意事項
a) 有機物及び濁りを含まない試料でニッケルの濃度が低い場合は,試料500 mLまでの適量をとり,6.1
を行い,B.11.2.3のa)〜h) に準じて操作して定量できる。この場合には,試料は前処理した全量を用
い,試薬はB.11.2.1のa)〜f) におけるものと同量を用いる。検量線は,試料と同様に操作して作成す
る。
b) 指示薬の代わりにリトマス試験紙を用いてもよい。
B.11.3
フレーム原子吸光法
試料を前処理した後,アセチレン-空気フレーム中に噴霧し,ニッケルによる原子吸光を波長232.0 nm
で測定してニッケルを定量する。
定量範囲:Ni:0.3〜6 mg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
B.11.3.1 試薬
試薬は,次による。
a) ニッケル標準液(Ni:10 mg/L) 24.7.1 e) のニッケル標準液(Ni:100 mg/L)10 mLを全量フラスコ
100 mLにとり,硝酸(1+1)[21.6.1 b) による。]2 mLを加え,水を標線まで加える。
B.11.3.2 器具及び装置
器具及び装置は,21.3.2 a) による。
B.11.3.3 準備操作
準備操作は,次による。
a) 試料を箇条6によって処理する。
B.11.3.4 操作
操作は,次による。
a) B.11.3.3の準備操作を行った試料を,JIS K 0121の8.(操作方法)に従って,フレーム中に噴霧し,
波長232.0 nmの指示値(吸光度又はその比例値)を読み取る。
b) 空試験として,B.11.3.3の準備操作での試料と同量の水をとり,試料と同様にB.11.3.3及びa) の操作
を行って試料について得た指示値を補正する。
c) あらかじめ,次によって作成したニッケルの検量線から,試料中のニッケルの濃度(mg/L)を算出す
194
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
る。
1) ニッケル標準液(Ni:10 mg/L)3〜60 mLを全量フラスコ100 mLに段階的にとり,B.11.3.3 a) を行
った試料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。この溶液についてa) 及
びb) の操作を行い,ニッケル(Ni)の濃度と指示値との関係線を作成する。検量線の作成は,試
料測定時に行う。
B.11.3.5 留意事項
a) 準備操作として溶媒抽出法を適用するときは,ニッケル標準液の量を,適宜,減らす。
b) ニッケルの濃度が低い試料で,抽出操作を妨害する物質を含まない場合は,次の操作を行い,ニッケ
ルの定量に用いてもよい。試料100 mLをビーカにとり,JIS K 8180で規定する塩酸5 mLを加え,約
5分間煮沸した後,放冷する。B.11.2.3のa)〜c) に準じて操作し,ニッケルをジメチルグリオキシム
錯体として,クロロホルム層に抽出する。クロロホルム層を合わせ,塩酸(1+20)[B.11.2.1 b) によ
る。]10 mLを加え,振り混ぜてニッケルを逆抽出する。水層を分離した後,クロロホルム層に塩酸(1
+20)5 mLを加えて逆抽出を行う。逆抽出液を合わせ,全量フラスコ25 mLに移し入れ,水を標線
まで加える。この溶液を用いて,ニッケルの定量を行う。
B.11.4
ICP発光分光分析法
24.7による。
B.12
アルミニウム(Al)
B.12.1 一般事項
アルミニウムの定量には,8-キノリノール吸光光度法,フレーム原子吸光法,電気加熱原子吸光法又は
ICP発光分光分析法を適用する。
B.12.2 8-キノリノール吸光光度法
微酸性にした試料に,塩化ヒドロキシルアンモニウムと1,10-フェナントロリンとを加えて鉄をマスキン
グした後,8-キノリノール及び酢酸アンモニウムを加え,生成する錯体をクロロホルムで抽出する。シア
ン化カリウムを含む塩化アンモニウム溶液で洗浄して,アルミニウムとともに抽出された銅,ニッケル,
コバルトなどを除去した後,アルミニウム錯体の吸光度を測定してアルミニウムを定量する。
定量範囲:Al:10〜100 μg/L(試料500 mLを使用する場合),繰返し精度:3〜10 %
B.12.2.1 試薬
試薬は,次による。
a) 水 21.2.1 a) による。
b) 塩酸(1+2) JIS K 8180で規定する塩酸を用いて調製する。
c) アンモニア水(1+2) JIS K 8085で規定するアンモニア水を用いて調製する。
d) 硫酸ナトリウム JIS K 8987で規定するもの。
e) 塩化ヒドロキシルアンモニウム溶液(100 g/L) 22.2.1 d) による。
f)
酢酸アンモニウム溶液(150 g/L) JIS K 8359で規定する酢酸アンモニウム15 gを水に溶かして100
mLとする。この溶液を分液漏斗に入れ,8-キノリノール-クロロホルム溶液(JIS K 8775で規定する
8-キノリノール2 gをJIS K 8322で規定するクロロホルム100 mLに溶かす。)5 mLを加え,激しく振
り混ぜた後,放置し,クロロホルム層を捨てる。この操作を,クロロホルム層が着色しなくなるまで
繰り返す。次に,水層にクロロホルム5 mLを加え,激しく振り混ぜた後,放置し,クロロホルム層
を捨てる。この操作を,水層に黄色が認められなくなるまで繰り返す。水層は乾いたろ紙5種Bでろ
195
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
過し,溶液中のクロロホルムの小滴を除く。
g) シアン化カリウム-塩化アンモニウム溶液 JIS K 8443で規定するシアン化カリウム1.0 gを水に溶か
して500 mLとする。これにJIS K 8116で規定する塩化アンモニウムを少量ずつ溶かし,pHを9.0〜
9.5に調節する。この溶液はf) の酢酸アンモニウム溶液(150 g/L)と同様に操作して8-キノリノール
クロロホルム溶液及びクロロホルムで洗浄し,精製する。
警告 シアン化カリウムは有毒である。取扱い及び廃液の処理は,法令に従い安全及び健康に対す
る適切な措置を取らなければならない。シアン化カリウムを含む溶液は,有害なシアン化水
素ガスが発生するため酸性にしてはならない。
h) 1,10-フェナントロリン溶液(1 g/L) 26.2.1 f) による。
i)
8-キノリノール溶液(10 g/L) JIS K 8775で規定する8-キノリノール2 gにJIS K 8355で規定する
酢酸5 mLを加え,僅かに加熱して溶かした後,水を加えて200 mLとする。
j)
クロロホルム JIS K 8322で規定するもの。
k) アルミニウム標準液(Al:100 mg/L) 国家計量標準にトレーサブルなアルミニウム標準液(Al:100
mg/L)を使用するか,又は,次による。JIS K 8255で規定する硫酸カリウムアルミニウム・12水[ビ
ス(硫酸)カリウムアルミニウム-水(1/12)]1.76 gをとり,塩酸(1+1)[21.5.1 b) による。]20 mL
に溶かし,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。または,JIS K 8069で規定する
アルミニウム(99.9 %以上)0.100 gをとり,塩酸(1+1)20 mL中に加熱して溶かし,放冷後,全量
フラスコ1 000 mLに移し入れ,水を標線まで加える。
l)
アルミニウム標準液(Al:1 mg/L) アルミニウム標準液(Al:100 mg/L)10 mLを全量フラスコ1 000
mLにとり,塩酸(1+1)[21.5.1 b) による。]20 mLを加え,水を標線まで加える。
B.12.2.2 器具及び装置
器具及び装置は,次による。
a) 分液漏斗 200 mL
b) 光度計 分光光度計又は光電光度計
B.12.2.3 操作
操作は,次による。
a) 箇条6の操作を行った試料の適量(Alとして5〜50 μgを含む。)をとり,塩化ヒドロキシルアンモニ
ウム溶液(100 g/L)1 mLと1,10-フェナントロリン溶液(1 g/L)5 mLとを加えて振り混ぜ,アンモニ
ア水(1+2)を滴加してpHを約3.5に調節する。pHの確認には,ブロモフェノールブルー試験紙を
用いるとよい。
b) 水を加えて液量を約80 mLとした後,約15分間放置する。
c) 8-キノリノール溶液(10 g/L)3 mLと酢酸アンモニウム溶液(150 g/L)10 mLとを加え,アンモニア
水(1+2)を滴加してpHを5.2〜5.5に調節する。pHの確認には,ブロモクレゾールグリーン試験紙
を用いるとよい。pHが5.2〜5.5の範囲にならないときは,塩酸(1+2)又はアンモニア水(1+2)を
用いて調節する。
d) この溶液を分液漏斗に移し,水を加えて液量を約100 mLとした後,クロロホルム10 mL(又は20 mL)
を加え,約1分間激しく振り混ぜて放置する。
e) クロロホルム層を分離して,別の分液漏斗に移し入れ,シアン化カリウム-塩化アンモニウム溶液25
mLを加え,振り混ぜて放置する。
f)
クロロホルム層を共栓試験管30 mLに入れ,硫酸ナトリウム約1 gを加えて軽く振り混ぜて水分を除
196
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
く。
g) クロロホルム層の一部を吸収セルに移し,クロロホルムを対照液として波長390 nm付近の吸光度を
測定する。
h) 空試験として水約70 mLをとり,a)〜g) の操作を行って吸光度を測定し,試料について得た吸光度を
補正する。
i)
あらかじめ,次によって作成したアルミニウムの検量線から,試料中のアルミニウムの濃度(mg/L)
を算出する。
1) アルミニウム標準液(Al:1 mg/L)5〜50 mLを分液漏斗に段階的にとり,水を加えて液量約70 mL
とし,a)〜h) の操作を行ってアルミニウムの濃度と吸光度との関係線を作成する。
B.12.2.4 留意事項
a) 前処理には箇条6のうち6.3の方法は用いない。有機物が少ない試料の場合には,試料100 mLにつき
JIS K 8180で規定する塩酸5 mLを加え,静かに加熱して液量が約1/5になるまで濃縮してもよい。
b) 試料にふっ化物イオンが含まれる場合には,ふっ化物イオン0.5 mgに対し硫酸ベリリウム36 mgを加
えておけば,妨害を防ぐことができる。
c) クロムが存在する場合には,pH 5.2〜5.5における抽出は,できるだけ低温で行う。氷水で冷却すると
よい。
d) マンガンが多量に含まれる場合には,錯体を抽出したクロロホルム溶液を,塩化ヒドロキシルアンモ
ニウムを加えたpH 7以下の酢酸-酢酸アンモニウム溶液(JIS K 8359で規定する酢酸アンモニウム173
gを水約800 mLに溶かし,この溶液にJIS K 8355で規定する酢酸13 mLを加え,水で1 Lとする。)
で洗浄してマンガンを除去する。
e) チタン,モリブデンなどが含まれている場合には,B.12.2.3 c) の操作で,銅,ニッケル,コバルトな
どを除いた後,pH 10のアンモニアアルカリ性塩化アンモニウム溶液(50 g/L)(JIS K 8116で規定す
る塩化アンモニウム50 gを水約500 mLに溶かし,この溶液にJIS K 8085で規定するアンモニア水を
pH約10まで加え,水で1 Lとする。)25 mLにJIS K 8230で規定する過酸化水素2 mLを加えた溶液
でクロロホルム層を洗浄する。
f)
この方法では,鉄0.45 mgまでの存在は影響しない。
B.12.3 フレーム原子吸光法
試料を前処理した後,アセチレン-一酸化二窒素フレーム中に噴霧し,アルミニウムによる原子吸光を波
長309.3 nmで測定してアルミニウムを定量する。
定量範囲:Al:5〜100 mg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
B.12.3.1 試薬
試薬は,次による。
a) 塩化カリウム溶液(100 g/L) JIS K 8121で規定する塩化カリウム10 gを水に溶かして100 mLとす
る。
b) アルミニウム標準液(Al:500 mg/L) B.12.2.1 k) に準じて調製する。
B.12.3.2 器具及び装置
器具及び装置は,21.3.2 a) による。
B.12.3.3 準備操作
準備操作は,次による。
a) 試料を箇条6によって処理する。
197
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
B.12.3.4 操作
操作は,次による。
a) B.12.3.3 a) の準備操作を行った試料の適量(Alとして0.5〜10 mgを含む。)を全量フラスコ100 mL
にとり,JIS K 8180で規定する塩酸1 mLを加え,水を標線まで加える。
b) この溶液50 mLを乾いたビーカにとり,塩化カリウム溶液(100 g/L)2 mLを加える。
c) b) の試料をアセチレン-一酸化二窒素フレーム中に導入し,波長309.3 nmにおける指示値(吸光度又
はその比例値)を読み取る。
d) 空試験としてB.12.3.3 a) の準備操作での試料と同量の水をとり,試料と同様にB.12.3.3 a) 及びa)〜
c) の操作を行って指示値を読み取り,試料について得た指示値を補正する。
e) あらかじめ,次によって作成したアルミニウムの検量線から,試料中のアルミニウムの濃度(mg/L)
を算出する。
1) アルミニウム標準液(Al:500 mg/L)1〜20 mLを全量フラスコ100 mLに段階的にとり,B.12.3.3 a)
を行った試料と同じ酸の濃度になるように酸及びJIS K 8180で規定する塩酸を加えた後,水を標線
まで加える。この溶液についてb) 及びc) の操作を行う。
2) 別に,空試験として水についてB.12.3.3 a) を行った試料と同じ酸の濃度になるように酸及び塩酸を
加えた後,b) 及びc) の操作を行って標準液について得た指示値を補正する。
3) アルミニウムの濃度と指示値との関係線を作成する。検量線の作成は,試料測定時に行う。
B.12.4 電気加熱原子吸光法
試料を前処理した後,電気加熱炉で原子化し,アルミニウムによる原子吸光を波長309.3 nmで測定して
アルミニウムを定量する。
定量範囲:Al:20〜200 μg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
B.12.4.1 試薬
試薬は,次による。
a) 水 21.2.1 a) による。
b) 硝酸(1+1) 21.6.1 b) による。
c) アルミニウム標準液(Al:1 mg/L) B.12.2.1 k) のアルミニウム標準液(Al:100 mg/L)5 mLを全量
フラスコ500 mLにとり,硝酸(1+1)10 mLを加え,水を標線まで加える。使用時に調製する。
B.12.4.2 器具及び装置
器具及び装置は,21.4.2 a) による。
B.12.4.3 準備操作
準備操作は,次による。
a) 試料を箇条6によって処理する。
B.12.4.4 操作
操作は,次による。
a) B.12.4.3 a) の準備操作を行った試料の一定量(例えば,10〜50 μL)をマイクロピペットで発熱体に注
入し,乾燥(100〜120 ℃,30〜40秒間)した後,灰化(600〜1 000 ℃,30〜40秒間)し,次に,原
子化(2 200〜3 000 ℃,3〜6秒間)し,波長309.3 nmの指示値(吸光度又はその比例値)を読み取
る。引き続いて少なくともこの操作を3回繰り返し,指示値が合うことを確認する。
b) 空試験としてB.12.4.3 a) の準備操作での試料と同量の水をとり,試料と同様にB.12.4.3 a) 及びa) の
操作を行い,試料について得た指示値を補正する。
198
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
c) あらかじめ,次によって作成したアルミニウムの検量線から,試料中のアルミニウムの濃度(μg/L)
を算出する。
1) アルミニウム標準液(Al:1 mg/L)2〜20 mLを全量フラスコ100 mLに段階的にとり,B.12.4.3 a) を
行った試料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。この溶液についてa)
の操作を行う。
2) 別に,空試験として,水についてB.12.4.3 a) を行った試料と同じ酸の濃度になるように酸を加えた
後,a) の操作を行って標準液について得た指示値を補正し,アルミニウムの濃度と指示値との関係
線を作成する。検量線の作成は,試料測定時に行う。
B.12.4.5 留意事項
a) 乾燥,灰化及び原子化の条件は,装置によって異なる。試料の注入量及び共存する塩類の濃度によっ
ても異なることがある。
B.12.5 ICP発光分光分析法
試料を前処理した後,試料導入部を通して誘導結合プラズマ中に噴霧し,アルミニウムによる発光を波
長309.271 nmで測定してアルミニウムを定量する。
定量範囲:50〜5 000 μg/L 繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
B.12.5.1 試薬
試薬は,次による。
a) 水 24.4.1 a) による。
b) 硝酸(1+1) 21.6.1 b) による。
c) アルミニウム標準液(Al:10 mg/L) B.12.2.1 k) のアルミニウム標準液(Al:100 mg/L)10 mLを全
量フラスコ100 mLにとり,硝酸(1+1)2 mLを加え,水を標線まで加える。使用時に調製する。
d) イットリウム溶液(Y:1 000 mg/L) 21.5.1 f) による。
e) イットリウム溶液(Y:50 mg/L) 21.5.1 g) による。
B.12.5.2 器具及び装置
器具及び装置は,次による。
a) ICP発光分光分析装置 JIS K 0116による。
B.12.5.3 準備操作
準備操作は,次による。
a) 試料を6.6によって前処理する。
b) 空試験として,試料と同量の水を用いてa) の操作を行う。
B.12.5.4 操作
操作は,次による。
a) B.12.5.3 a) の溶液をJIS K 0116の4.7(定量分析)に準じて,試料導入部を通してプラズマ中に噴霧
し,波長309.271 nmの発光強度を測定する。
b) 空試験としてB.12.5.3 b) についてa) の操作を行って,試料について得た発光強度を補正する。
c) あらかじめ,次によって作成したアルミニウムの検量線から,試料中のアルミニウムの濃度(mg/L又
はμg/L)を算出する。
1) アルミニウム標準液(Al:10 mg/L)を全量フラスコ100 mLに測定濃度範囲を含むように,アルミ
ニウム標準液を段階的にとり,B.12.5.3 a) 試料と同じ酸の濃度となるように硝酸(1+1)を加えた
後,水を標線まで加える。これらの溶液についてa) の操作を行う。
199
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2) 別に,空試験としてこの操作に用いた水について試料と同じ条件になるように硝酸(1+1)を加え
た後,b) の操作を行ってアルミニウム標準液について得た発光強度を補正し,アルミニウムの濃度
と発光強度との関係線を作成する。検量線の作成は試料測定時に行う。
B.12.5.5 留意事項
a) 波長の異なる2本以上のスペクトル線を同時測定が可能な装置では,内標準法によることができる。
1) 内標準法を用いるときは,B.12.5.3 a) で処理した試料の適量を全量フラスコ100 mLにとり,B.12.5.1
e) のイットリウム溶液(Y:50 mg/L)10 mLを加え,B.12.5.3 a) の試料と同じ酸の濃度になるよう
に酸を加えた後,水を標線まで加える。
2) この溶液についてB.12.5.4 a) の操作を行って測定対象元素の波長と同時に371.029 nm(イットリウ
ム)の発光強度を測定し,アルミニウムとイットリウムとの発光強度の比を求める。
3) 空試験として試料と同量の水を用いて,1) 及び2) の操作を行う。
4) あらかじめ,次によって作成した各測定対象元素の検量線から,試料中の測定対象元素の濃度を算
出する。
4.1) アルミニウム標準液(Al:10 mg/L)を全量フラスコ100 mLに測定濃度範囲を含むように,アル
ミニウム標準液を段階的にとり,B.12.5.1 e) のイットリウム溶液(Y:50 mg/L)10 mLをそれぞ
れ加え,B.12.5.3 a) の試料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。こ
れらの溶液についてB.12.5.4 a) の噴霧操作を行ってアルミニウムの濃度におけるアルミニウムと
イットリウムとの発光強度比を求める。
4.2) 別に空試験として混合標準液に代えて水を用い,同様に操作・測定を行ってアルミニウムとイッ
トリウムとの発光強度比を求め,アルミニウムの発光強度比を補正する。検量線の作成は試料測
定時に行う。
200
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書C
(参考)
脱ガス酸電気伝導率の測定
C.1 脱ガス酸電気伝導率
揮発性物質処理を行っているボイラにおいて,給水及び蒸気の酸電気伝導率を測定する場合に,起動時
の二酸化炭素の侵入又は使用薬品の種類によっては,酸電気伝導率の値が二酸化炭素の影響を受けて高く
なる場合がある。給水及び蒸気中の塩類の存在を正しく知るためには,図C.1の測定原理[1]に示すように
試料水を陽イオン交換後に,加熱沸騰方式又は脱気膜によって溶存する二酸化酸素を除去して,酸電気伝
導率を測定する必要がある。
C.1.1 試薬
試薬は,次による。
a) 強酸性陽イオン交換樹脂 次のように水素イオン形に変換したものを用いる。
強酸性陽イオン交換樹脂の適量を水(イオン交換樹脂量の約3倍量)に浸し,12時間以上放置した
後,適切なカラム(イオン交換樹脂を充塡したとき,イオン交換樹脂層の高さが約600 mmになるよ
うなカラム)に水で移し入れる。強酸性陽イオン交換樹脂1 Lにつき8.3.1 a) の塩酸(1+11)20 Lを,
8〜10 L/h・(L-樹脂) で流し,引き続き水3 Lを同じ流量で流して水素イオン形に変換する。次に,水
を約20 L/h・(L-樹脂) の割合で80 L/(L-樹脂) 以上流して洗浄する。これを図4のイオン交換カラムに
気泡が入らないように水で充塡する。
C.1.2 器具及び装置
器具及び装置は,次による。
a) 陽イオン交換-脱ガス装置 強酸性陽イオン交換カラムと二酸化酸素を除去する脱ガス装置で構成さ
れる。陽イオン交換-脱ガス装置の一例を図C.2[2]及び図C.3[3]に示す。図C.2に示す装置は,試料水を
陽イオン交換後に加熱し,沸騰させることによって二酸化炭素などの溶存するガスを除去するもので
あり,陽イオン交換カラム,脱ガス装置(蒸気発生ユニット,熱交換器)で構成する。図C.3に示す
装置は,試料水を陽イオン交換後に脱気膜に通水し,真空ポンプで二酸化炭素などの溶存するガスを
除去するものであり,イオン交換カラム,脱ガス装置(脱気膜,ポンプユニット)で構成する。
b) 電気伝導率計 電気伝導率0.01〜1 mS/m(0.1〜10 μS/cm)(25 ℃)を測定できるもので,セル定数0.8
〜12 m−1(0.008〜0.12 cm−1)の流液形検出器が使用できるもの。
c) 温度計(熱電対) 許容差±1 ℃のもの。JIS C 1602で規定する熱電対を用いる。
C.1.3 準備操作
脱ガス酸電気伝導率プロセス用分析装置が安定に達した後,次による。
a) スパン校正 校正用電気抵抗で指示値を所定の値に調整する。
C.1.4 操作
操作は,次による。
a) 陽イオン交換-脱ガス装置内の水素イオン形に変換した強酸性陽イオン交換樹脂を充塡したイオン交
換カラムに試料を12〜15 L/h・(L-樹脂) の割合で連続的に流す。
b) a) の流出液を脱ガス装置に導入し,二酸化炭素などの溶存ガスを除去する。
c) b) の流出液を流液形検出器に導入し,電気伝導率を測定する。
201
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
C.1.5 留意事項
強酸性陽イオン交換樹脂の代わりに,カチオン交換膜を用いた電気式カチオン交換器を使用してもよい。
C.2 脱ガス酸電気伝導率プロセス用分析装置による測定方法
加熱沸騰方式又は脱気膜方式の脱ガス酸電気伝導率プロセス用分析装置を用いて,給水,蒸気などに含
まれる二酸化炭素を除去した後の酸電気伝導率を連続的に測定する。
測定範囲:0.002〜500 mS/m(0.02〜5 000 μS/cm),繰返し精度:±1 %以下。
C.2.1 試薬
試薬は,次による。
a) 強酸性陽イオン交換樹脂 C.1.1 a) による。これをイオン交換カラムに気泡が入らないように水で充
塡する。
C.2.2 器具及び装置
器具及び装置は,次による。
a) 脱ガス酸電気伝導率プロセス用分析装置 箇条3 a) で規定するプロセス用分析装置で,イオン交換カ
ラム,脱ガス装置,検出部,指示部,記録部で構成する。その構成の一例を図C.4に示す。酸電気伝
導率が1 mS/m(10 μS/cm)以下の場合には,JIS K 0552で規定する二重温度補償ができる方式のもの
を用いる。
C.2.3 準備操作
脱ガス酸電気伝導率プロセス用分析装置の校正は,C.1.3による。
C.2.4 操作
脱ガス酸電気伝導率プロセス用分析装置の操作は,C.1.4による。
C.2.5 点検・整備
点検・整備は定期的に行うこととし,次による。
a) 試料の流量
b) セル(検出部)の汚れ
c) 温度補償素子の点検
d) 陽イオン交換樹脂の再生
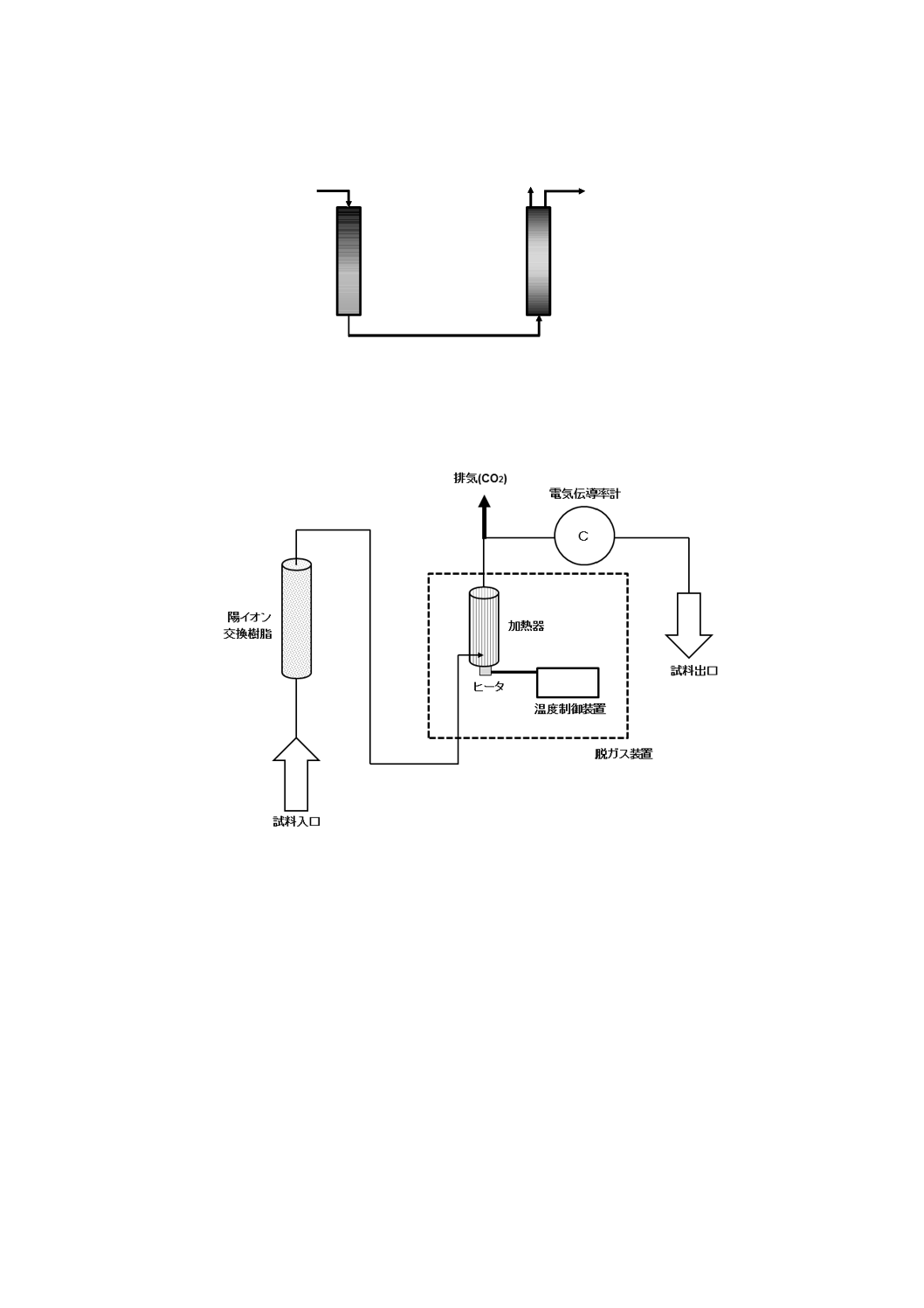
202
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
脱ガス酸電気伝導率
電気伝導率
酸電気伝導率
脱ガス装置
陽イオン交換カラム
H+, Cl-, OH-
CO2
NH4+, OH-
Na+, H+, Cl-,
HCO3-
H+, Cl-, CO2, HCO3-, OH-
R−NH4+
R−Na+
R−H+
図C.1−脱ガス酸電気伝導率測定の原理
図C.2−脱ガス酸電気伝導率装置の一例(加熱沸騰による脱ガス方式)
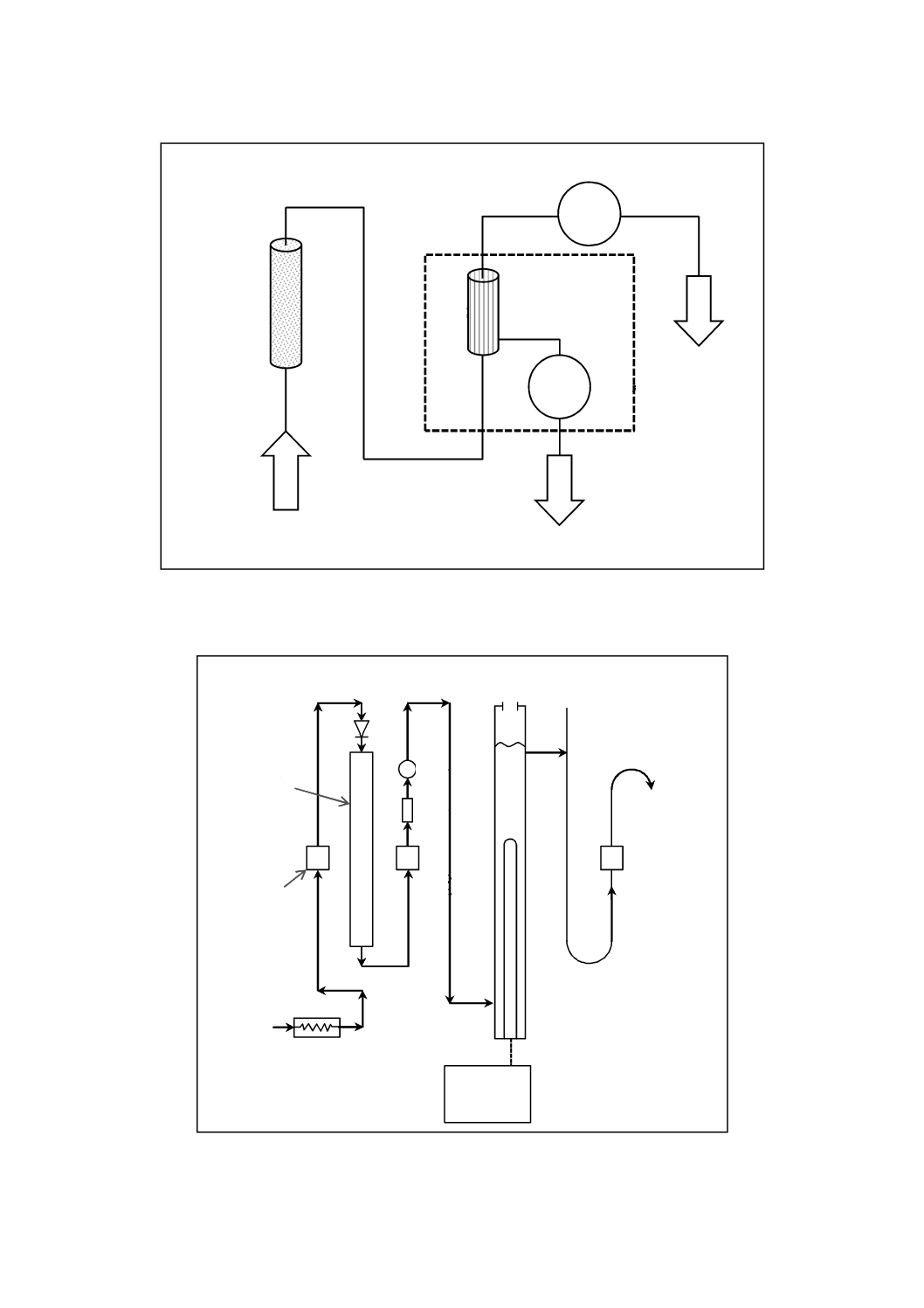
203
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図C.3−脱ガス酸電気伝導率装置の一例(脱気膜による脱ガス方式)
図C.4−脱ガス酸電気伝導率装置の一例
陽イオン
交換樹脂
電気伝導率計
脱気膜
真空ポンプ
脱ガス装置
C
P
試料入口
試料出口
排気
試料出口
脱ガス
酸電気伝導率計
セル
熱交換器
電気伝導率計
セル
温度計
流量計
酸電気伝導
率計セル
試料入口
陽イオン交換
カラム
加熱器
排気
(脱気ガス)
排気
(脱気ガス)
温度制御装置

204
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書D
(参考)
腐食電位及び酸化還元電位の測定
高入力インピーダンス電位差計によって給水及び復水中などにおいて,プラントの測定対象箇所の腐食
電位及び酸化還元電位は,測定対象箇所を構成している材料と同じ素材(高圧・高温配管用の炭素鋼)を
用いて測定対象箇所の温度に近い温度で連続的に測定する。ただし,高温高圧用銀-塩化銀内部電極及び高
温高圧用銀-塩化銀外部電極の最高使用温度が300 ℃程度であるので,測定対象箇所は,300 ℃以下の給
水系及び復水系とする。
測定範囲:±1 000 mV{銀-塩化銀電極[塩化カリウム溶液(0.1 mol/L)]}
D.1 試薬
試薬は,次による。
a) 水 JIS K 0557で規定するA2又はA3の水。試薬の調製及び操作には,この水を用いる。
b) 塩化カリウム溶液(0.1 mol/L) JIS K 8121で規定する塩化カリウム(電気伝導率測定用)約8 gを
500 ℃で約4時間加熱し,デシケータ中で放冷する。その7.455 gをはかりとり,少量の水に溶かし,
全量フラスコ1 000 mLに移し入れ,水を標線まで加える。この溶液は,参照電極の内部液に用いる。
D.2 器具及び装置
器具及び装置は,次による。
a) 参照電極 腐食電位及び酸化還元電位の測定に用いる,高温高圧用銀-塩化銀内部参照電極及び高温高
圧用銀-塩化銀外部参照電極の一例を,それぞれ図D.1及び図D.2に示す。いずれの電極の場合も通常,
内部液にはD.1 b) の塩化カリウム溶液(0.1 mol/L)又は0.01 mol/Lの同溶液を用いる。
図D.1−高温高圧用銀-塩化銀内部参照電極の一例(圧力容器の内側に設置)
205
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図D.2−高温高圧用銀-塩化銀外部参照電極の一例(圧力容器の外側に設置)
b) 指示電極
1) 白金電極 酸化還元電位測定用の白金電極は,次による。
白金電極は,白金板(約20×20 mmの厚さ約1 mm)を図D.3に示すように加工し,白金電極部
以外のリード線部などには,熱収縮四ふっ化エチレン樹脂製のチューブで被覆し,加熱して熱収縮
させたもの。使用前にJIS K 8034で規定するアセトンで洗浄し,D.1 a) の水で十分に洗浄した後,
測定に用いる。
単位 mm
図D.3−酸化還元電位測定用の白金電極の一例
206
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2) 試料電極 腐食電位測定用の試料電極は,次による。
試料電極(炭素鋼電極)は,プラントの測定対象箇所を構成している材料と同じ素材の炭素鋼管
(高温・高圧配管用)を約20×20 mm,厚さ約3 mmに切り取り,JIS R 6252で規定する研磨紙の
粒度P 800までのものを用いて乾式で仕上げ研磨し,図D.4に示すように加工し,試料電極部以外
のリード線部などには,熱収縮四ふっ化エチレン樹脂製のチューブで被覆し,加熱して熱収縮させ
たもの。使用前にJIS K 8034で規定するアセトンで洗浄し,D.1 a) の水で十分に洗浄した後,測定
に用いる。
単位 mm
図D.4−腐食電位測定用の試料電極の一例
c) 温度計(熱電対) JIS C 1602で規定する熱電対のN又はKのそれぞれクラス1(温度測定範囲−40 ℃
以上375 ℃未満)を用いる。
d) 圧力容器 試料採取部の圧力より数MPa程度高い最高使用圧力をもつ圧力容器(オートクレーブ)を
用いる。
e) 高入力インピーダンス電位差計 オートクレーブ対応形の高入力インピーダンス電位差計を用いる。
なお,高入力インピーダンス電位差計の内部抵抗は,1 TΩ(1×1012 Ω)以上のものを用いる。キャ
ピラリを高温高圧用銀-塩化銀外部電極に接続するには,接続するキャピラリのインピーダンスが1
GΩ(1×109 Ω)程度以下になるようにする。
f)
腐食電位及び酸化還元電位の計測系統 電位計測系統は,高入力インピーダンス電位差計の計測部を
はじめとして,試料採取装置,電極部,冷却器,減圧装置(圧力調節弁),及び記録部で構成する。そ
の構成例を図D.5に示す。
なお,電極部は,参照電極,白金電極,試料電極及び温度計(熱電対)を備えた圧力容器で構成す
る。その構成例を図D.6及び図D.7に示す。
207
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図D.5−電位計測系統の構成の一例
図D.6−内部参照電極を用いた腐食電位及び酸化還元電位計測用の電極部の構成例
試料採取装置
電極部
計測部
記録部
冷却器
減圧装置
排出水
試料電極
白金電極
電位差計
圧力容器(オートクレーブ)
四ふっ化エチレン樹脂
被覆リード線
試験水出口
試験水入口
内部参照電極
耐圧シール
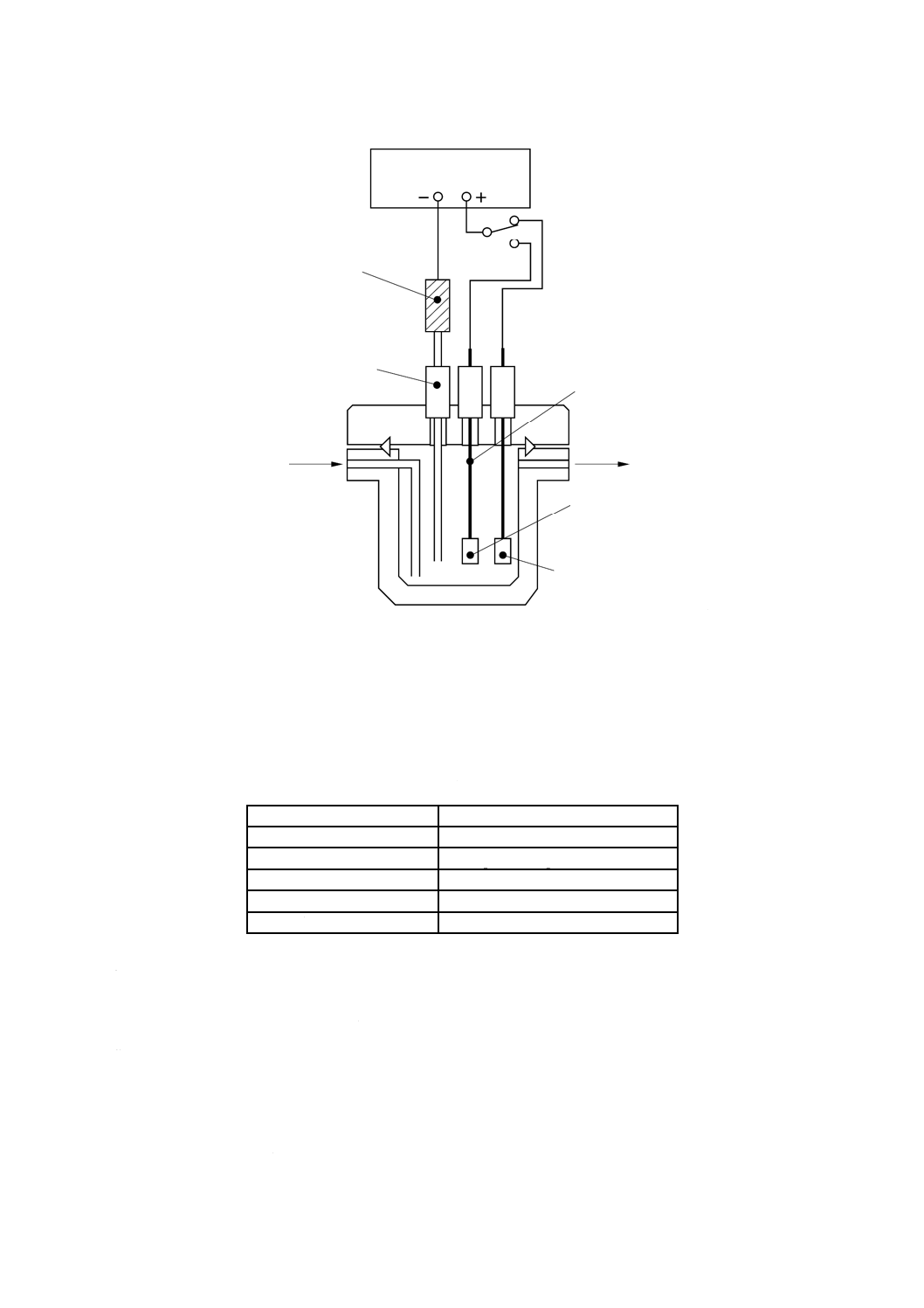
208
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図D.7−外部参照電極を用いた腐食電位及び酸化還元電位計測用の電極部の構成例
また,腐食電位及び酸化還元電位の測定に用いる高入力インピーダンス電位差計の仕様の一例を,
表D.1に示す。
表D.1−高入力インピーダンス電位差計の仕様の一例
測定範囲
±10 V
内部抵抗
1 TΩ以上(T:テラ1012)
漏れ電流
1 pA以下(p:ピコ10−12)
入力方式
差動入力,又はシングルエンド方式
入出力間の絶縁の有無
あり
測定精度
±0.5 %(フルスケール)
D.3 測定
測定は,次による。
a) 校正 校正は,高入力インピーダンス電位差計が安定状態に達した後,次の操作を行う。
1) 電位差計の校正 電位差計に基準電圧を印加して所定の指示値が得られることを確認し,誤差があ
る場合には調整する。
2) 参照電極の校正 高温高圧用銀-塩化銀内部参照電極及び高温高圧用銀-塩化銀外部参照電極が,正
常な電位をもつことを確認するため,銀-塩化銀[塩化カリウム溶液(飽和)]電極を用いて検定す
る。すなわち,濃度0.1 mol/L,又は0.01 mol/Lの塩化カリウム溶液中で,銀-塩化銀[塩化カリウム
電位差計
試験水出口
試験水入口
外部参照電極
耐圧シール
四ふっ化エチレン樹脂
被覆リード線
圧力容器(オートクレーブ)
試料電極
白金電極
209
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
溶液(飽和)]電極に対する高温高圧用銀-塩化銀内部参照電極及び高温高圧用銀-塩化銀外部参照電
極の電位を室温で測定する。この室温における電位の値は,0.1 mol/Lの塩化カリウム内部液の場合
には,15 ℃で82±10 mV,20 ℃で85±10 mV,25 ℃で88±10 mV,30 ℃で91±10 mVであり,
0.01 mol/Lの塩化カリウム内部液の場合には,15 ℃で140±10 mV,20 ℃で143±10 mV,25 ℃で
147±10 mV,30 ℃で151±10 mVである[1]。室温での測定値がこの範囲内に入らない場合には,参
照電極の内部液を交換する。内部液の交換によっても電位が所定の値を示さない場合には,塩化銀
の再生,又は銀-塩化銀電極の交換を行う。
なお,0.01 mol/Lの塩化カリウム内部液の場合には,高温における塩化銀の溶解によって塩化物
イオン濃度が僅かに変化しても大きな電位変動が起こるので[2],0.1 mol/Lの塩化カリウム内部液を
用いる方が望ましい。
b) 測定準備 電位計測系統の各部を点検し,特に,水漏れのないことを確認した後,所定の手順に従っ
て試料採取装置によって所定の圧力まで減圧した試料(給水,復水など)を電極部に流す。この場合
に,検出部を構成する耐圧容器内での沸騰を防止するため,圧力容器に流入する試料温度を測定し,
その温度に相当する飽和圧力[蒸気表(温度基準)[3]]から求める。]より1 MPa程度高い圧力になる
ように,試料の圧力を試料採取装置の減圧装置(圧力調節弁)で調節する。
例えば,試料採取装置の入口における試料温度が300 ℃,圧力が20 MPaの場合,蒸気表から300 ℃
の飽和圧力は8.587 MPaであるので,圧力容器の圧力を約9.6 MPa(8.587+1=9.587≒9.6 MPa)にな
るように試料採取装置の減圧装置(圧力調節弁)で調節する。
c) 測定 校正が終了したら,試料を所定の流量で流し,指示値が安定した後,連続測定を行う。
D.4 点検・整備
点検・整備は,定期的に行うこととし,次による。
a) 試料の流量
b) 参照電極,試料電極及び白金電極の汚染
c) 参照電極の内部液の補充
d) 記録データの採取
D.5 標準水素電極(SHE)基準電位への換算
高温高圧用銀-塩化銀内部参照電極及び高温高圧用銀-塩化銀外部参照電極を用いて測定した電位を,標
準水素電極(SHE)基準の電位に換算するための換算値(加算値)を表D.2に示す。すなわち,高温高圧
用銀-塩化銀内部参照電極及び高温高圧用銀-塩化銀外部参照電極を用いて測定した電位に表D.2に示した
電位を加算する。
210
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表D.2−高温高圧用銀-塩化銀参照電極で測定した電位をSHEの電位に
換算するための換算値(加算値)(0.1 mol/Lの塩化カリウム内部液の場合)
温度
(℃)
銀-塩化銀内部参照電極電位
(mV)
銀-塩化銀外部参照電極電位
(mV)(室温25 ℃の場合)
25
50
100
150
200
250
300
282
269
235
188
130
59.5
−22.6
287
262
211
158
101
36
−38
なお,参考までに0.1 mol/Lの塩化カリウム内部液の高温高圧用銀-塩化銀内部参照電極及び高温高圧用
銀-塩化銀外部参照電極を用いて測定した電位を,標準水素電極(SHE)基準の電位に換算するための式を,
それぞれ式(D.1)[4]及び式(D.2)[5][6]に示す。
内部参照電極の場合
ESHE(t)(mV)=Eobs+2.38×102−5.38×10−1 t−2.37×10−3 t2+1.99×10−1 (t+273.15) ········· (D.1)
ここで,Eobs:測定電位,t:試験水温度(℃)
外部参照電極の場合
ESHE(T)(mV)=Eobs+286.6−1.003 ΔT+1.745×10−4 ΔT 2−3.030×10−6 ΔT 3 ····················· (D.2)
ここで,Eobs:測定電位,ΔT=T−t,T:試験水温度(℃),t:室温(℃)
211
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考文献
(附属書A,附属書B及び附属書C関係)
JIS G 3458 配管用合金鋼鋼管
JIS K 0410-3-7 水質−サンプリング−第7部:ボイラ施設の水及び蒸気のサンプリングの指針
JIS K 0801 濁度自動計測器
JIS K 8069 アルミニウム(試薬)
JIS K 8122 塩化カルシウム二水和物(試薬)
JIS K 8123 塩化カルシウム(試薬)
JIS K 8136 塩化すず(II)二水和物(試薬)
JIS K 8247 過マンガン酸カリウム(試薬)
JIS K 8272 キシレンシアノールFF(試薬)
JIS K 8312 クロム酸カリウム(試薬)
JIS K 8397 サリチル酸ナトリウム(試薬)
JIS K 8488 1,5-ジフェニルカルボノヒドラジド(試薬)
JIS K 8498 ジメチルグリオキシム(試薬)
JIS K 8517 二クロム酸カリウム(試薬)
JIS K 8528 しゅう酸ナトリウム(試薬)
JIS K 8529 臭素(試薬)
JIS K 8545 硝酸アンモニウム(試薬)
JIS K 8580 すず(試薬)
JIS K 8722 ペンタシアノニトロシル鉄(III)酸ナトリウム二水和物(試薬)
JIS K 8775 8-キノリノール(試薬)
JIS K 8785 二りん酸ナトリウム十水和物(試薬)
JIS K 8798 フェノール(試薬)
JIS K 8842 ブロモチモールブルー(試薬)
JIS K 8844 ブロモフェノールブルー(試薬)
JIS K 8847 ヘキサメチレンテトラミン(試薬)
JIS K 8872 ホルムアルデヒド液(試薬)
JIS K 8992 硫酸ヒドラジニウム(試薬)
JIS R 6252 研磨紙
(附属書B関係)
[1] Standard Methods for the Examination of Water and Wastewater, 1998, 20th Edition, pp4-132 and 4-133.
[2] BENSON, B.B and KRAUSE, D. jr. 1980. The concentration and isotopic fractionation of gases dissolved in
fresh water in equilibrium with the atmosphere: I. Oxygen. Limnol. Oceanogr. 25:662.
[3] MORTIMER, C.H. 1981. The oxygen content of air-saturated fresh waters over ranges of temperature and
atmospheric pressure of limnological interest. Int. Assoc. Theoret. Appl. Limnol., Communication No. 22,
Stuttgart, West Germany.
212
B 8224:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
[4] BENSON, B.B. and KRAUSE, D. jr. 1984. The concentration and isotopic fractionation of oxygen dissolved in
freshwater and seawater in equilibrium with the atmosphere. Limnol. Oceanogr. 29:620.
(附属書C関係)
[1] David M.Gray, Power Plant Chemistry 2005,7(4), 214
[2] D4519-94 Standard Test Method for On-Line Determination of Anions and Carbon Dioxide in High Purity
Water by Cation Exchange and Degassed Cation Conductivity
[3] 日機装株式会社技術資料
(附属書D関係)
[1] 東伸工業株式会社技術資料
[2] 腐食防食協会編(1986):防食技術便覧,p.1077,日刊工業新聞社
[3] 日本機械学会編(1999):蒸気表
[4] R.S.Greely, W.T.Smith, R.W.Stoughton and M.H.Lietke:J.Phys.Chem., Vol.64, p.652 (1960)
[5] Digby.D.Macdonald, Arthur.C.Scott, and Paul Wentrcek:“External Reference Electrodes for Use in High
Temperature Aqueous Systems”, J. Electrochem. Soc., Vol.126, No.6,p.908-911 (1979)
[6] 橘 孝二:“酸化ジルカロイ管を用いた高温高圧水用圧力平衡型外部照合電極の試作と分極測定への応
用”,防食技術,Vol.34, No.2, p125-131 (1985)