T 0801:2016 (ISO 11135:2014)
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
目 次
ページ
序文 ··································································································································· 1
1 適用範囲························································································································· 1
1.1 適用 ···························································································································· 1
1.2 適用除外 ······················································································································ 2
2 引用規格························································································································· 3
3 用語及び定義 ··················································································································· 3
4 品質マネジメントシステム ································································································ 11
4.1 文書化 ························································································································ 11
4.2 経営者の責任 ··············································································································· 11
4.3 製品実現 ····················································································································· 11
4.4 測定,分析及び改善−不適合製品の管理············································································ 11
5 滅菌剤の特性 ·················································································································· 11
5.1 一般 ··························································································································· 11
5.2 滅菌剤 ························································································································ 12
5.3 微生物殺滅効果の有効性 ································································································ 12
5.4 材料への影響 ··············································································································· 12
5.5 安全性及び環境 ············································································································ 12
6 プロセス及び装置の特性 ··································································································· 12
6.1 一般 ··························································································································· 12
6.2 プロセスの特性 ············································································································ 12
6.3 装置の特性 ·················································································································· 13
7 製品の決定(Product definition) ······················································································· 14
7.1 一般 ··························································································································· 14
7.2 製品の安全性,品質及び性能 ·························································································· 14
7.3 微生物学的品質 ············································································································ 14
7.4 文書化 ························································································································ 15
8 プロセスの決定(Process definition) ·················································································· 15
9 バリデーション ··············································································································· 15
9.1 一般 ··························································································································· 15
9.2 据付適格性の確認(IQ) ································································································ 16
9.3 運転適格性の確認(OQ) ······························································································· 16
9.4 稼働性能適格性の確認(PQ) ························································································· 17
9.5 バリデーションのレビュー及び承認·················································································· 18
10 日常監視及び管理 ·········································································································· 20
11 滅菌からの製品のリリース ······························································································ 21
T 0801:2016 (ISO 11135:2014) 目次
(2)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ページ
12 プロセス有効性の維持 ···································································································· 22
12.1 一般 ·························································································································· 22
12.2 装置のメンテナンス ····································································································· 22
12.3 適格性の再確認 ··········································································································· 22
12.4 変更の評価 ················································································································· 22
12.5 同等性の評価 ·············································································································· 23
附属書A(規定)滅菌プロセスの致死率の決定−バイオロジカルインジケータ/バイオバーデン法 ····· 24
附属書B(規定)安全率を見込んだ滅菌プロセスの致死率の決定−オーバーキル法 ·························· 25
附属書C(参考)温度センサ,湿度センサ及びバイオロジカルインジケータの数 ····························· 26
附属書D(参考)規定要求事項の適用に関する指針 ··································································· 29
附属書E(規定)単一ロットの出荷 ························································································ 67
参考文献 ···························································································································· 69
T 0801:2016 (ISO 11135:2014)
(3)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法第12条第1項の規定に基づき,一般社団法人日本医療機器学会(JSMI)及
び一般財団法人日本規格協会(JSA)から,工業標準原案を具して日本工業規格を制定すべきとの申出が
あり,日本工業標準調査会の審議を経て,厚生労働大臣が制定した日本工業規格である。
これによって,JIS T 0801-1:2010は廃止され,この規格に置き換えられた。
この規格は,著作権法で保護対象となっている著作物である。
この規格の一部が,特許権,出願公開後の特許出願又は実用新案権に抵触する可能性があることに注意
を喚起する。厚生労働大臣及び日本工業標準調査会は,このような特許権,出願公開後の特許出願及び実
用新案権に関わる確認について,責任はもたない。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
T 0801:2016
(ISO 11135:2014)
ヘルスケア製品の滅菌−エチレンオキサイド−
医療機器の滅菌プロセスの開発,
バリデーション及び日常管理の要求事項
Sterilization of health care products-Ethylene oxide-
Requirements for the development, validation and routine control of
a sterilization process for medical devices
序文
この規格は,2014年に第2版として発行されたISO 11135を基に,技術的内容及び構成を変更すること
なく作成した日本工業規格である。
なお,この規格で点線の下線を施してある参考事項は,対応国際規格にはない事項である。
1
適用範囲
1.1
適用
この規格は,産業界及びヘルスケア施設の両者における医療機器のエチレンオキサイド滅菌のプロセス
の開発,バリデーション及び日常管理の要求事項について規定する。
なお,この規格への適用については,両者において類似点又は相違点があっても差し支えない。
注記1 類似点は品質システム,職員の教育及び適切な安全に関わる手順についての共通な必要性で
ある。主な相違点は,ヘルスケア施設における独特な物理的及び組織の状況に関係し,更に,
滅菌前の再使用可能医療機器の初期状態に関係する。
注記2 ヘルスケア施設は,処理エリアの物理的設計,用いる装置,及び適切な訓練,経験の水準に
ついての職員の能力が医療機器製造業者とは異なる。ヘルスケア施設の最も大切な機能は患
者の看護であり,医療機器の再生処理は患者の看護業務をサポートする作業の一部にすぎな
い。
注記3 滅菌前の医療機器の初期状態について,医療機器製造業者では一般的に,バージン材料で製
造した同一で大量の医療機器を滅菌する。一方,ヘルスケア施設では,新規の医療機器及び
各種の形態,並びに種々のバイオバーデンレベルの再使用可能医療機器の取扱い及び再生処
理をしなければならない。よってヘルスケア施設の担当者は医療機器の洗浄,洗浄の評価,
組付け及び包装といった滅菌前の追加の課題に直面しなければならない。この規格ではヘル
スケア施設に独自な代替えの方法及び指針を医療機器製造業者のそれとは別に示している。
注記4 熱及び/又は湿気に敏感で湿熱滅菌ができない医療機器について,エチレンオキサイド(EO)
ガス及びその混合物は,第一選択肢として使用される有効な滅菌剤である。
注記5 この規格の適用範囲は医療機器に限定しているが,ここで規定する要求事項及び提供する指
2
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
針は,その他のヘルスケア製品に適用できる。
注記6 この規格の対応国際規格及びその対応の程度を表す記号を,次に示す。
ISO 11135:2014,Sterilization of health-care products−Ethylene oxide−Requirements for the
development, validation and routine control of a sterilization process for medical devices(IDT)
なお,対応の程度を表す記号“IDT”は,ISO/IEC Guide 21-1に基づき,“一致している”
ことを示す。
1.2
適用除外
1.2.1
この規格は,スクレイピー,牛海綿状脳症及びクロイツフェルト・ヤコブ病のような海綿状脳症病
原物質の不活化プロセスの開発,バリデーション及び日常管理の要求事項については規定しない。クロイ
ツフェルト・ヤコブ病などの発症因子で汚染された危険性のある材料を処理するのには,日本では固有の
勧告がある。
注記 平成9年4月24日薬機第71号厚生労働省薬務局医療機器開発課長通知“クロイツフェルト・
ヤコブ病感染防止のための医療用具の消毒について”参照。
1.2.2
この規格は,医療機器を“無菌”と表示するための特定の要求事項の詳細は規定しない。
注記 医療機器に“無菌”と表示するための国又は地域の規制要求事項に注意を払うことが必要であ
る。例えば,EN 556-1又はANSI/AAMI ST67を参照。
1.2.3
この規格は,医療機器を製造する全てのプロセスを管理するための品質マネジメントシステムを規
定するものではない。
注記 定義し文書化した手順の効果的な実施は,医療機器の滅菌プロセスの開発,バリデーション及
び日常管理に必要である。このような手順は一般的に品質マネジメントシステムの要素と考え
られる。製造又は再生処理において,完全な品質システムを構築することはこの規格の要求事
項ではない。必要とされる品質マネジメントシステムの要素は,規格の文書中の適切な箇所に
規定として参照している(特に,箇条4参照)。医療機器の製造又は再生処理の全ての段階を管
理する品質マネジメントシステムについての規格(JIS Q 13485を参照)に注意を払うとよい。
医療機器の提供に対する国及び/又は地域の規制では,完全な品質マネジメントシステムの実
施及び第三者機関によるその品質マネジメントシステムの評価を要求する場合がある。
1.2.4
この規格は,労働安全に関わるEO滅菌施設の設計及び運転についての要求事項は規定しない。
注記1 労働安全についての詳細情報については参考文献を参照すること。国の規制が存在すること
に注意する。
注記2 EOは毒性があり,可燃性であり爆発性がある。EOの取扱い及びEOを使用する作業所に対
して安全要求事項を提供する規制が存在することに注意する。
1.2.5
この規格は,個々の包装容器又はフレキシブルチャンバにEO又はその混合物を直接導入する滅菌
法には適用しない。
注記 これらのタイプのEOプロセスについては,ISO 14937参照。
1.2.6
この規格はEO残留及び/又はその反応生成物の量の測定法には適用しない。
注記1 詳細な情報はJIS T 0993-7を参照。
注記2 医療機器の上又は医療機器内のEO残留物のレベルの限度値を規定する国の規制が存在する
ことに注意を払うのがよい。
3
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2
引用規格
次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成する。これらの
引用規格のうちで,西暦年を付記してあるものは,記載の年の版を適用し,その後の改正版(追補を含む。)
は適用しない。西暦年の付記がない引用規格は,その最新版(追補を含む。)を適用する。
JIS Q 10012 計測マネジメントシステム−測定プロセス及び測定機器に関する要求事項
注記 対応国際規格:ISO 10012,Measurement management systems−Requirements for measurement
processes and measuring equipment(IDT)
JIS Q 13485:2005 医療機器−品質マネジメントシステム−規制目的のための要求事項
注記 対応国際規格:ISO 13485:2003,Medical devices−Quality management systems−Requirements
for regulatory purposes(IDT)
JIS T 0993-7 医療機器の生物学的評価−第7部:エチレンオキサイド滅菌残留物
注記 対応国際規格:ISO 10993-7,Biological evaluation of medical devices−Part 7: Ethylene oxide
sterilization residuals及びTechnical Corrigendum 1:2009(IDT)
JIS T 11737-1 医療機器の滅菌−微生物学的方法−第1部:製品上の微生物群の測定方法
注記 対応国際規格:ISO 11737-1,Sterilization of medical devices−Microbiological methods−Part 1:
Determination of a population of microorganisms on products(IDT)
JIS T 11737-2 医療機器の滅菌−微生物学的方法−第2部:滅菌プロセスの定義,バリデーション及
び維持において実施する無菌性の試験
注記 対応国際規格:ISO 11737-2,Sterilization of medical devices−Microbiological methods−Part 2:
Tests of sterility performed in the definition, validation and maintenance of a sterilization process
(IDT)
ISO 11138-1:2006,Sterilization of health care products−Biological indicators−Part 1: General requirements
ISO 11138-2:2006,Sterilization of health care products−Biological indicators−Part 2: Biological indicators
for ethylene oxide sterilization processes
ISO 11140-1,Sterilization of health care products−Chemical indicators−Part 1: General requirements
3
用語及び定義
この規格で用いる主な用語及び定義は,次による。
3.1
エアレーション(aeration)
滅菌プロセスの一部で,エチレンオキサイド及び/又はその反応生成物を,あらかじめ決めたレベルに
達するまで,医療機器から脱離する操作。
注記 滅菌器の中及び/又はそれとは別のチャンバ又は部屋において実施できる。
3.2
エアレーションエリア(aeration area)
エアレーションを行うチャンバ又は部屋。
3.3
バイオバーデン(bioburden)
製品及び/又は無菌バリアシステムの上又は内部に存在する生育可能な微生物群。
(ISO/TS 11139:2006の定義2.2参照)
4
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3.4
バイオロジカルインジケータ,BI(biological indicator,BI)
ある特定の滅菌プロセスに対して,あらかじめ定めた抵抗性を示す生育可能な微生物を含む試験システ
ム。
(ISO/TS 11139:2006の定義2.3参照)
3.5
校正(calibration)
計器若しくは測定系の示す値,又は実量器若しくは標準物質の表す値と,標準によって実現される値と
の間の関係を確定する一連の作業。
(ISO/TS 11139:2006の定義2.4参照)
3.6
ケミカルインジケータ(chemical indicator)
プロセスにばく(曝)露することで生じる化学的又は物理的な変化に基づき,あらかじめ定義した一つ
又は複数の滅菌プロセス変数の変化を表すシステム。
(ISO/TS 11139:2006の定義2.6参照)
3.7
コンディショニング(conditioning)
エチレンオキサイドの導入前に,製品をあらかじめ定めた温度及び相対湿度に到達させるための処理。
この処理は滅菌サイクルに含まれる。
注記1 滅菌サイクル中のこの部分は,大気圧又は減圧下で実施することができる。
注記2 3.27のプレコンディショニング参照。
3.8
D値(D value)
D10値(D10 value)
定められた条件下で,試験に用いる微生物数の90 %を不活性化するのに要する時間又は線量。
(ISO/TS 11139:2006の定義2.11参照)
注記 この規格では,D値は試験微生物の菌数を90 %不活性化するのに必要とするばく露時間のこと
である。
3.9
開発(development)
仕様を作り上げる行為。
(ISO/TS 11139:2006の定義2.13参照)
3.10
露点(dew point)
飽和水蒸気圧がその大気での水蒸気分圧に等しい温度。
注記 大気が露点より低い温度に冷却すると水の凝縮が起こる可能性がある。
3.11
確立(establish)
理論的評価によって決定し,実験によって確認すること。
(ISO/TS 11139:2006の定義2.17参照)
5
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3.12
エチレンオキサイド導入時間,EO導入時間(ethylene oxide injection time)
EO(EO混合物)のチャンバへの導入開始から終了するまでの時間。
3.13
ばく(曝)露時間(exposure time)
プロセスパラメータを,それぞれにあらかじめ定めた許容範囲内に維持した時間。
(ISO/TS 11139:2006の定義2.18参照)
注記 サイクル致死率の計算では,EOの導入を終了してからEOの除去が始まるまでの滅菌サイクル
の時間。
3.14
許容外(fault)
あらかじめ定めた許容範囲から一つ又は複数のプロセスパラメータが外れること。
(ISO/TS 11139:2006の定義2.19参照)
3.15
フラッシング(flushing)
チャンバへのろ過した空気,不活性ガス又は蒸気の導入及び抜気とを交互に繰り返すこと,又はろ過し
た空気,不活性ガス又は蒸気を滅菌負荷及び滅菌チャンバに連続的に通すことによって滅菌負荷及び滅菌
チャンバからエチレンオキサイドを除去する手順。
3.16
部分サイクル(fractional cycle)
あらかじめ定めた滅菌プロセスに対して,ばく露時間を減少させたサイクル。
3.17
ハーフサイクル(half cycle)
あらかじめ定めた滅菌プロセスに対して,ばく露時間を50 %に減少させたサイクル。
3.18
ヘルスケア施設,HCF(health care facility,HCF)
健康の増進及び維持並びに傷病の防止並びに処置を実施する公的又は民間の組織及び公共施設。
例 ヘルスケア施設には病院,療養施設,長期介護施設,独立外来外科センター,医院,診療所又は
歯科医院がある。
3.19
ヘルスケア製品[health care product (s)]
体外診断用医療機器を含む医療機器,又は生物製剤を含む医薬品。
(ISO/TS 11139:2006の定義2.20参照)
3.20
据付適格性の確認,IQ(installation qualification,IQ)
装置がその要求仕様に適合して提供され,かつ,据え付けられたことの証拠を取得し,文書化するプロ
セス。
(ISO/TS 11139:2006の定義2.22参照)
3.21
医療機器(medical device)
6
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
あらゆる計器,器械,用具,機械,器具,埋込み用具,体外診断薬,検定物質,ソフトウェア,材料又
はその他の同類のものは関連する物質であって,単独使用か組合せ使用かを問わず,製造業者が人体への
使用を意図し,その使用目的が次の一つ以上である。
− 疾病の診断,予防,監視,治療又は緩和
− 負傷の診断,監視,治療,緩和又は補助
− 解剖学的又は生理学的なプロセスの検査,代替又は修復
− 生命支援又は維持
− 受胎調整
− 医療機器の殺菌
− 人体から採取される標本の体外試験法による医療目的のための情報提供
薬学,免疫学又は新陳代謝の手段によって体内又は体表において意図したその主機能を達成することは
ないが,それらの手段によって機能の実現を補助するもの。
(JIS Q 13485:2005の定義3.7参照)
3.22
微生物(microorganism)
細菌,真菌,原虫及びウイルスを包含する微小体。
注記 ある種の規格によっては,滅菌プロセスのバリデーション及び/又は日常管理において,上記
で定義した全てのタイプの微生物の不活化の滅菌プロセスの有効性を立証することを要求しな
いこともある。
(ISO/TS 11139:2006の定義2.26参照)
3.23
運転適格性の確認,OQ(operational qualification,OQ)
据え付けられた装置をその操作手順に従って用いたとき,あらかじめ定めた限度内で作動する証拠を取
得し,文書化するプロセス。
(ISO/TS 11139:2006の定義2.27参照)
3.24
オーバーキル法(overkill approach)
製品のバイオバーデンと同等以上の抵抗性のあるバイオロジカルインジケータに対して,少なくとも,
12芽胞対数減少(SLR)を与える滅菌プロセスを用いる方法。
3.25
パラメトリックリリース(parametric release)
プロセスパラメータがあらかじめ定めた許容範囲内で運転されたことを証明する記録に基づいて,製品
が滅菌済みであると宣言すること。
(ISO/TS 11139:2006の定義2.29参照)
注記 このプロセスからのリリースの方法では,バイオロジカルインジケータを使用しない。
3.26
稼働性能適格性の確認,PQ(performance qualification,PQ)
操作手順に従って据え付けられ,運転されている装置が,あらかじめ定めた判断基準に恒常的に適合し
て稼働し,その結果,仕様に適合する製品を生産することができるという証拠を取得し,文書化するプロ
セス。
7
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(ISO/TS 11139:2006の定義2.30参照)
3.27
プレコンディショニング(preconditioning)
部屋又はチャンバ内で,滅菌サイクル前に,製品をあらかじめ定めた温度及び相対湿度に到達させるた
めの処理。
3.28
プロセスチャレンジデバイス,PCD(process challenge device,PCD)
滅菌プロセスに対し,定義した抵抗性を示すように設計された,滅菌プロセスの性能を,評価するため
に用いられるもの。
(ISO/TS 11139:2006の定義2.33参照)
注記1 この規格では,PCDは微生物を直接又は間接に接種した製品,模擬製品,その他の機器であ
る。7.1.6及びD.7.1.6を参照。
注記2 この規格では,内部PCDと外部PCDとの区別がされている。内部PCDは,要求される製品
のSALが達成されたことを示すのに使用される。製品の中,製品の間又は製品搬送容器に置
かれたPCDは,内部PCDである。一方,搬送容器の間又は製品の外部の表面に置かれたPCD
は,外部PCDである。外部PCDは,日常の製造サイクルでの微生物学的監視に使用する目
的で設計されたものである。
3.29
プロセスパラメータ(process parameter)
あらかじめ定めたプロセス変数の値。
(ISO/TS 11139:2006の定義2.34参照)
注記 滅菌プロセスの仕様には,プロセスパラメータ及びその許容範囲が含まれる。
3.30
プロセス変数(process variable)
滅菌プロセスの条件で,その変化が微生物の殺滅効果に変動を与えるような条件。
例えば,時間,温度,圧力,濃度,湿度,波長。
(ISO/TS 11139:2006の定義2.35参照)
3.31
処理カテゴリ(processing category)
同一の条件で滅菌できる違った種類の製品又は製品ファミリの集まり。
注記 このカテゴリに含まれる全ての製品は,この集まりに対するプロセスチャレンジデバイスより
も滅菌プロセスに対して抵抗性が同等か低いことを示すことによって決定される。
3.32
製品(product)
プロセスの結果。
(JIS Q 9000:2006の定義3.4.2参照)
注記 この規格では,製品は有形のものであり,原料,中間品,半組立品及びヘルスケア製品でもあ
り得る。
3.33
製品ファミリ(product family)
8
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
定義したプロセス条件を用いて滅菌が可能であるプロセス特性をもつ製品のグループ。
3.34
製品負荷容積(product load volume)
製品が占有できる有効チャンバ容積内の定められた空間。
3.35
公的微生物保存機関(recognized culture collection)
特許手続上の微生物寄託の国際的承認に関するブタペスト条約に基づく国際的保存機関。
(ISO/TS 11139:2006の定義2.38参照)
3.36
標準菌(reference microorganism)
公的微生物保存機関から得られる菌株。
(ISO/TS 11139:2006の定義2.39参照)
3.37
適格性の再確認(requalification)
定義した滅菌プロセスが引き続き許容できるものであることを確認するために,バリデーションの一部
分を反復実施すること。
(ISO/TS 11139:2006の定義2.40参照)
3.38
再使用可能医療機器(reusable medical device)
医療機器製造業者が再生処理及び再使用に適したように指定又は意図した医療機器。
注記 医療機器製造業者が,単回使用を指定又は意図した医療機器ではない。
3.39
サービス(services)
外部から供給を受けるもので,装置が正常な機能を発揮するのに必要なもの。
例えば,電気,水,圧縮空気,排水。
(ISO/TS 11139:2006の定義2.41参照)
3.40
単回使用医療機器(single use medical device)
医療機器製造業者が,1回限りの使用を指定又は意図した医療機器。
3.41
あらかじめ定める(specify)
承認を受けた文書の中で詳細を明記すること。
(ISO/TS 11139:2006の定義2.42参照)
3.42
芽胞対数減少,SLR(Spore-Log-Reduction,SLR)
最初の芽胞の菌数N0の対数から最後の芽胞の菌数NUの対数を引いた数。
(ISO 14161:2009の定義3.19参照)
注記 あらかじめ定めた条件下でのばく露によるバイオロジカルインジケータ又は接種したものの上
の芽胞数の減少について示す。
9
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
直接計数法では,
u
0log
log
SLR
N
N−
=
ここに,
N0: 最初の菌数
Nu: 最後の菌数
フラクションネガティブ法では,
−
=
n
q
N
ln
log
log
SLR
0
ここに,
N0: 最初の菌数
q: 試験したサンプル数
n: 菌の生育を示さなかったサンプルの数
インジケータの生残が認められなかった場合,正確なSLRの計算ができない。1個のインジケータの生
残が確認された場合,SLRはlog N0より大きいと報告することができる。
3.43
無菌(sterile)
生育可能な微生物が存在しないこと。
(ISO/TS 11139:2006の定義2.43参照)
3.44
無菌バリアシステム(sterile barrier system)
微生物の侵入の防止及び使用時点での製品の無菌提供を可能にする最低限の包装。
(ISO/TS 11139:2006の定義2.44参照)
3.45
無菌性(sterility)
生育可能な微生物が存在しない状態。
注記1 実際には,そのような微生物の存在しない絶対的な状態を証明することはできない。
注記2 3.47滅菌を参照。
(ISO/TS 11139:2006の定義2.45参照)
3.46
無菌性保証水準,SAL(sterility assurance level,SAL)
滅菌後に,生育可能な1個の微生物が製品上に存在する確率。
注記 SALは定量値として一般的に,10−3又は10−6と表す。この定量値を無菌性保証に適用すると
きは,10−6のSALは10−3のSALよりも小さい値であるがより高い無菌性保証を与える。
(ISO/TS 11139:2006の定義2.46参照)
3.47
滅菌(sterilization)
製品を生育可能な微生物が存在しない状態にするために用いる,バリデートされたプロセス。
注記1 滅菌プロセスでは,微生物の不活化は指数関数で表現される。したがって,個々の製品に生
残する生育可能な微生物は,確率論の観点から表現できる。この確率は,非常に低い数に減
らすことはできるが,決してゼロに減らすことはできない。
注記2 3.46無菌性保証水準を参照。
(ISO/TS 11139:2006の定義2.47参照)
10
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3.48
滅菌サイクル(sterilization cycle)
密閉された滅菌チャンバ内で実施する,脱気,コンディショニング(行う場合),エチレンオキサイド,
不活性ガス(行う場合)導入,エチレンオキサイドによるばく露,エチレンオキサイドの除去,並びにフ
ラッシング(行う場合)及び空気又は不活性ガスの追加からなる処理。
3.49
滅菌負荷(sterilization load)
滅菌プロセスを用いて一緒に滅菌される,又は滅菌された製品。
(ISO/TS 11139:2006の定義2.48参照)
3.50
滅菌プロセス(sterilization process)
あらかじめ定めた無菌性についての要求事項を達成するための一連の活動又は操作。
(ISO/TS 11139:2006の定義2.49参照)
注記 この一連の活動又は操作には,あらかじめ定めた条件でのプレコンディショニング(必要な場
合),エチレンオキサイドヘのばく露及びエチレンオキサイド並びにその副生成物除去に必要な
全ての後処理を含む。これには滅菌プロセスに先立つ全ての洗浄,消毒又は包装操作は含まな
い。
3.51
滅菌専門家(sterilization specialist)
利用する滅菌技術,材料への影響及び微生物について技術的知識をもつ者。
3.52
滅菌剤(sterilizing agent)
あらかじめ定めた条件下で,無菌性を達成するために十分な殺菌作用をもつ物理的若しくは化学的媒体
又はその組合せ。
(ISO/TS 11139:2006の定義2.50参照)
3.53
生残曲線(survivor curve)
定めた条件下での滅菌剤へのばく露の増加に対応する微生物数の不活化を図式的に表現したもの。
(ISO/TS 11139:2006の定義2.51参照)
3.54
無菌試験(test for sterility)
最終プロセスを経た製品に対して実施する薬局方で定義された技術的操作。
(ISO/TS 11139:2006の定義2.53参照)
3.55
無菌性の試験(test of sterility)
開発,バリデーション又は適格性の再確認の一部として実施する技術的操作で,製品又はその一部に生
育可能な微生物の存在の有無を判定するために行う試験。
(ISO/TS 11139:2006定義の2.54参照)
3.56
有効チャンバ容積(usable chamber volume)
11
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
固定部品又は可動部品によって制限されない,滅菌負荷を入れることができる滅菌チャンバ内の使用で
きるあらかじめ定めた空間。
注記 チャンバ内のガス循環のための空間は有効容積には含まない。
3.57
バリデーション(validation)
プロセスが,恒常的にあらかじめ決められた仕様に適合する製品が得られることを確立するために,要
求される結果を得て,記録し及び解釈するための文書化した手順。
(ISO/TS 11139:2006の定義2.55参照)
3.58
バージン材料(virgin material)
使用されていないか,又はその材料の製造以外の処理をしていない材料。
4
品質マネジメントシステム
4.1
文書化
4.1.1
開発,バリデーション,日常管理及び滅菌からの製品リリースの手順をあらかじめ定めなければな
らない。
4.1.2
この規格が要求する文書及び記録は,あらかじめ指名した職員(4.2.1参照)によってレビューし,
承認しなければならない。文書及び記録は,JIS Q 13485の該当する箇条によって管理しなければならない。
4.2
経営者の責任
4.2.1
この規格の要求事項を実施し,これに適合するための責任及び権限をあらかじめ定めなければなら
ない。責任は,JIS Q 13485の該当する箇条によって,力量のある職員に割り当てなければならない。
4.2.2
この規格の要求事項を,他の品質マネジメントシステムの組織によって実行する場合は,それぞれ
の組織の責任及び権限をあらかじめ定めなければならない。
ヘルスケア施設が再使用可能医療機器の滅菌を外部委託する場合,バリデーションと滅菌した製品のリ
リースはヘルスケア施設の責任である。
4.3
製品実現
4.3.1
購買の手順をあらかじめ定めなければならない。これらの手順は,JIS Q 13485の該当する箇条に
適合しなければならない。
4.3.2
製品の識別及びトレーサビリティの手順をあらかじめ定めなければならない。これらの手順は,JIS
Q 13485の該当する箇条に適合しなければならない。
4.3.3
この規格の要求事項に適合するために,試験用の計器を含む全ての機器の校正について,JIS Q
13485又はJIS Q 10012の該当する箇条に適合したシステムを構築しなければならない。
4.4
測定,分析及び改善−不適合製品の管理
不適合と認定した製品の管理,修正,是正処置及び予防処置の手順をあらかじめ定めなければならない。
これらの手順は,JIS Q 13485の該当する箇条に適合しなければならない。
5
滅菌剤の特性
5.1
一般
この箇条の目的は,滅菌剤を特定し,その微生物殺滅効果を立証し,微生物殺滅効果に影響する因子を
識別し,滅菌剤へのばく露が材料に与える影響を評価し,職員の安全及び環境保護への要求事項を明確に
12
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
することである。この活動は,試験又は試作システムで実施してもよい。その場合,最終的な装置の仕様
(6.3参照)は,試験又は試作システムで実施した実験的研究の結果に関連付けなければならない。この規
格において滅菌剤はEOである。
5.2
滅菌剤
滅菌剤の仕様には,該当する場合,有効期間でEOを仕様の範囲内に維持できる貯蔵条件を含めなけれ
ばならない。
5.3
微生物殺滅効果の有効性
一般に認められた組成外のEO又は新規の希釈剤を使用する場合は,これらの微生物殺滅効果の有効性
についてのデータを作成(開発)しておかなければならない。
注記 EOの微生物の不活化については多くの文献がある。これらの文献には,微生物の不活化に影
響するプロセス変数についての情報が記載されている。ただし,この規格ではこのような微生
物不活化についての一般的な研究を参照することは要求しない。
5.4
材料への影響
医療機器の製造に用いる様々な材料に対してEOが与える影響については,多くの文献があり,これら
の文献は,EOで滅菌される医療機器の設計及び開発に携わる者に有用である。この規格では,材料に及
ぼす影響についての個別の検討を行うことは要求しないが,製品自体に対するEOの影響については,検
討を行うことを要求している(箇条7参照)。
5.5
安全性及び環境
5.5.1
EO及びその希釈剤(ある場合)の安全データシート(SDS)又は類似の安全性情報を利用できる
ようにしなければならない。職員の健康及び安全を保護する必要な手段を明確にしなければならない。
5.5.2
滅菌プロセスを実施することによって環境が被る潜在的な影響について評価するとともに,環境を
保護する方法を明確にしなければならない。潜在的な影響,管理するための方法を含んだこの評価は文書
化しなければならない。
5.5.3
EOの使用者は,EO及びその希釈物並びに副生成物の放出及び廃棄については,国,地方及び国
際的な要求事項に従わなければならない。
6
プロセス及び装置の特性
6.1
一般
6.1.1
この箇条の目的は,滅菌プロセスを安全に再現性よく運用するために必要な滅菌プロセス全体及び
装置を定義することである。
6.1.2
既存のプロセスを製品の滅菌に使用する場合,この箇条の内容は必要ではないが,そのプロセス及
び装置は,6.2及び6.3で識別した変数が,日常の生産のプロセス仕様に含まれていることを確実にするた
めに,レビューをするとよい。
6.2
プロセスの特性
6.2.1
プロセスの特性には,少なくとも次の事項を含めなければならない。
a) EO滅菌プロセスに必要なフェーズの識別
b) 各フェーズのプロセス変数の識別
c) プロセス変数の文書化
注記 製品の定義(箇条7参照)で開発されたデータは滅菌プロセスの特性に影響することがある。
6.2.2
滅菌プロセスのフェーズには次を含む。
13
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) プレコンディショニング(行う場合)
b) 滅菌サイクル
c) エアレーション(行う場合)
6.2.3
プレコンディショニング(行う場合)のプロセス変数には少なくとも次を含む。
a) 時間
b) 温度
c) 湿度
d) 移送時間
6.2.4
滅菌サイクルのプロセス変数には次を含む。
a) ばく露時間
b) 温度
c) 湿度
d) EO濃度
e) 圧力
6.2.5
エアレーション(行う場合)のプロセス変数には少なくとも次を含む。
a) 時間
b) 温度
注記 エアレーションでのこれらのパラメータは,滅菌プロセスの微生物殺滅効果を確実にするため
にエアレーションを行う場合にだけプロセス変数として考慮される。
(AAMI TIR16:2009の5.1.3.3参照)
6.3
装置の特性
6.3.1
使用する装置の仕様を開発し,文書化しなければならない。この仕様は次の項目を含めなければな
らない。
a) プレコンディショニングエリア(行う場合)
b) 滅菌装置
c) エアレーションエリア(行う場合)
注記 装置設計のある部分は,法令及び規制,又は他の規格による影響を受けることがある。
6.3.2
仕様には,次の事項を少なくとも含めなければならない。
a) 全ての必要な附属設備とともに,構成部材の材質を含む装置についての記載
b) チャンバへ滅菌剤を供給する方法についての記載
c) 蒸気を含む全てのガスをチャンバへ供給する方法についての記載
d) センサの特性及びその配置を含む滅菌プロセスを監視,制御及び記録するための計装装置の記載
e) 滅菌器によって認識される許容外
f)
職員及び環境の保護などの安全機能
g) 必要なサービス及び排出の制御に必要とされる仕様を含む据付要求事項
6.3.3
プロセスの管理及び/又は監視に用いるソフトウェアは,ソフトウェアがその設計仕様に適合して
いることを示す文書化した証拠を提供できるように品質システムの要素に従って作成し,バリデートしな
ければならない。
注記 ソフトウェアに関する追加の情報は,ISO/IEC 90003に注意を払うとよい。
6.3.4
プロセス変数を監視し制御する手段をあらかじめ定めなければならない。
14
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6.3.5
制御機能の不全の場合でも,無効なプロセスを有効と判定するようなプロセス変数の誤記録が起こ
らないような手段を設けておかなければならない。
注記 上記の手段は,制御及び監視を独立のシステムとする又は制御及び監視をクロスチェックして,
全ての不一致及び許容外を判別することによって達成できる場合がある。
7
製品の決定(Product definition)
7.1
一般
7.1.1
この箇条の目的は,滅菌前の製品の微生物学的品質及び製品の包装,滅菌に供する方法などを含ん
だ滅菌する製品を決定することである。
7.1.2
新規に設定したか若しくは一部を変更した製品,包装,又は載荷形態を導入する場合は,これに先
立って,製品の決定を実施しなければならない。以前にバリデートした製品,包装又は載荷形態との同等
性(滅菌プロセスへのチャレンジを参照することで)の立証は,製品の決定の必要事項を満たしたと考え
てよい。全ての同等性の立証は文書化しなければならない。
7.1.3
製品は,滅菌プロセス中に,空気の除去(該当する場合)ができ,滅菌プロセス中に熱,湿気及び
EOが浸透でき,更に,プロセスの最後にEOの除去ができるように設計しなければならない。
7.1.4
包装は,滅菌プロセスの間,空気の除去ができ,熱,湿気及びEOが浸透でき,更に,プロセスの
最後にEOの除去ができるように設計しなければならない。
7.1.5
載荷形態は,滅菌プロセスの間,空気の除去ができ,熱,湿気及びEOが浸透でき,更に,プロセ
スの最後にEOの除去ができるように設計しなければならない。
7.1.6
製品の最も滅菌しにくい部位においても,あらかじめ定めた滅菌プロセスが有効であることを立証
しなければならない。この立証は,新規の製品のプロセスの決定及びバリデーションを実施することでで
きる。又は,新規の製品と以前にバリデートした製品,若しくはあらかじめ定めた滅菌プロセスにばく露
した条件で,製品のSALを確認するのに使用した内部プロセスチャレンジデバイス(内部PCD)との同
等性を立証することで達成できる。
7.2
製品の安全性,品質及び性能
7.2.1
製品/包装に大きな影響を与える特定したプロセスパラメータの許容値を使用した滅菌プロセス
を適用した後でも,製品及びその包装が,安全性,品質及び性能についてあらかじめ定めた要求事項に適
合していることを確認しなければならない。
注記 設計管理は,JIS T 14971に記載されている一側面である。
7.2.2
複数回の滅菌サイクルを認める場合は,このようなプロセスが,製品及びその包装へ与える影響を
評価しなければならない。
7.2.3
滅菌プロセスにばく露した後の製品の生物学的安全性は,JIS T 0993規格群及びISO 10993規格群
の該当する部に従って確立しなければならない。
7.2.4
処理した製品がJIS T 0993-7の要求事項に適合するような,EO残留量を減じる手段を確立しなけ
ればならない。
7.3
微生物学的品質
7.3.1
滅菌操作に供する製品の微生物学的品質及び清浄度が管理されており,かつ,滅菌プロセスの有効
性を損なわないようにするためのシステムをあらかじめ定め,維持しなければならない。
注記 エンドトキシンはEOプロセスで破壊されない。エンドトキシン試験の指針はANSI/AAMI
ST72及び該当する薬局方に示されている。
15
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7.3.2
単回使用医療機器の場合は,定めた間隔でJIS T 11737-1によるバイオバーデンの推定を実施しな
ければならない。再使用可能医療機器については,定めた洗浄方法及び該当する場合は消毒プロセスの有
効性の評価をしなければならない。
注記 再滅菌可能医療機器の再生処理のために提供される情報に対する要求事項は,ISO 17664に規
定されている。洗浄及び消毒プロセスの有効性の評価の情報はISO 15883規格群の該当するパ
ートに示されている。
7.4
文書化
医療機器の製造業者は,製品の決定の結果を文書化しなければならない。
8
プロセスの決定(Process definition)
8.1
この箇条の目的は,決定(開発)した製品(箇条7参照)の滅菌について,バリデーション検討に
適用可能なプロセスの詳細な仕様を決定することである。
8.2
決定した製品に適用できる滅菌プロセスを確立しなければならない。決定した製品には,新規若し
くは変更した製品,包装,又は載荷形態を含む。
8.3
プロセスの決定の作業は,据付適格性の確認(IQ)及び運転適格性の確認(OQ)が完了した滅菌チ
ャンバ(研究用チャンバ又は製造用チャンバ)で実施しなければならない(9.2及び9.3参照)。
8.4
プロセスの特性で決定したプロセスパラメータ及び関連するプロセス変数の有効性は,文書化及び
記録で妥当性を示さなければなければならない(6.2参照)。
8.5
微生物学的チャレンジのあらかじめ定めた滅菌サイクルにおける微生物不活化速度は,附属書A若
しくは附属書Bに規定した方法,又は要求される無菌性保証水準(SAL)を立証する他の方法のうちの一
つを用いて決定しなければならない。
8.6
滅菌プロセスの確立の一部で用いるBIは,次の事項に適合するものでなければならない。
a) ISO 11138-2:2006の箇条5及び9.5に適合する。
b) EOに対して,滅菌する製品のバイオバーデンと少なくとも同等の抵抗性を示す。
c) 適切なPCDの中に配置する。
プロセスの決定,バリデーション,又は日常監視及び管理に使用するPCDの適切さを立証しなければな
らない。PCDは,製品内の最も滅菌が困難な部位でのバイオバーデンよりも滅菌に対して同等以上のチャ
レンジを示さなければならない。
注記 BIの選定,使用及び解釈の情報は,ISO 14161を参照。
8.7
滅菌プロセスの設定に用いる市販されているBIは,8.6及びISO 11138-1の該当する箇条の要求事項
に適合したものでなければならない。
8.8
ケミカルインジケータ(CI)を滅菌プロセスの決定の一部として使用する場合,ISO 11140-1に適合
しなければならない。
CIを滅菌プロセス確立の唯一の方法としてはならない。また,要求するSALが達成されたことの指標
として用いてはならない。
8.9
無菌性の試験を滅菌プロセスの確立のために用いる場合は,JIS T 11737-2に適合しなければならな
い。
9
バリデーション
9.1
一般
16
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9.1.1
バリデーションの目的は,プロセスの決定(箇条8参照)で確立した滅菌プロセスが有効であり,
再現性よく滅菌負荷内の製品へ提供できることを立証することである。バリデーションは,据付適格性の
確認(IQ),運転適格性の確認(OQ)及び稼働性能適格性の確認(PQ)の段階で構成する。手順及び/又
は計画書が承認されるまで試験を開始してはならない。
9.1.2
IQは滅菌器及び全ての附属機器が,仕様どおりに供給され,据え付けられたことを立証するため
に実施する。
9.1.3
OQは滅菌器が設計仕様の性能要求事項に適合する能力を立証するために実施する。
9.1.4
PQは製品を使用して滅菌器があらかじめ定めた基準に従って定常的に運転し,プロセスが無菌で
あり,あらかじめ定めた要求事項に適合する製品を製造することを立証するバリデーションの段階である。
IQ及びOQは,滅菌プロセスに用いる一つの装置について1回実施するだけでよい。PQはプロセスが
識別された受入基準に適合し,製品に対して要求するSALを達成できる能力をもっていることを立証する
ために,各々のバリデートする新規のプロセス及び/又は製品について実施するとよい。
9.2
据付適格性の確認(IQ)
9.2.1
装置
9.2.1.1
全ての附属装置を含む滅菌プロセスに使用する装置は,その設計仕様に適合しなければならな
い。
9.2.1.2
滅菌器は該当する安全規格に適合しなければならない。
9.2.1.3
装置の操作手順を定めなければならない。これらの操作手順には,次を含むが,これらに限定し
ない。
a) 具体的な運転方法
b) 許容外の状態及びそれを表示する方式,並びにそれに対して取るべき対応
c) 保守及び校正の方法
d) 技術サポートのための連絡先
9.2.2
据付適格性の確認
9.2.2.1
装置及び付随するサービスの据付けは,建築及び技術的図面に従わなければならない。据付けは
関連する全ての国,地域及び地方の規制に適合しなければならない。
9.2.2.2
据付けの方法は,あらかじめ定めなければならない。その中には,職員の健康及び安全に対する
適切な指示を含まなければならない。
9.2.2.3
仕様の範囲内でEOの品質及び組成の維持を確実にするための安全な貯蔵条件をあらかじめ定め
なければならない。
9.2.2.4
IQの実施前に,IQの間に使用する全ての試験計器の校正状況を確認しなければならない。
9.2.2.5
装置,配管及びその他の附属機器の図面は,据え付けた状態をIQの間に反映し,確定しなけれ
ばならない。
9.2.2.6
IQの間にシステムに加えられた変更は,設計及びプロセス仕様への影響を評価し,設計履歴ファ
イルに文書化しなければならない。
9.3
運転適格性の確認(OQ)
9.3.1
OQの実施前に,滅菌プロセスの監視,制御,表示又は記録(全ての試験計器を含む。)に使用さ
れる全ての計器が校正済みであることを確認しなければならない(4.3.3参照)。
9.3.2
OQは,据え付けた装置が,その運転仕様に合致する能力があることを立証しなければならない。
17
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9.4
稼働性能適格性の確認(PQ)
9.4.1
一般
9.4.1.1
PQは微生物学的及び物理的適格性の確認からなり,その製品を滅菌するのに使用する装置で実
施する。
9.4.1.2
新規若しくは変更した製品,包装,載荷形態,滅菌装置又は滅菌プロセスパラメータを導入する
に当たっては,以前にバリデートした製品,包装,載荷形態,装置又はプロセスとの同等性が文書化され
ていない場合は,PQを実施しなければならない(7.1.2,7.1.6及び12.5参照)。
9.4.1.3
PQは滅菌器が許容基準に従って定常的に稼働し,そのプロセスが意図したSALとなる製品を製
造できることを立証するために,PQは日常的に滅菌する製品又は材料を代表するものを用いて実施しな
ければならない。
9.4.1.4
製品の載荷形態など,製品の滅菌への供給方法をあらかじめ定めなければならない。
注記 バリデーションを販売する製品を用いて行う場合は,7.2で患者への使用に当たっての製品品質
に関する情報を提供している。また,11.4では,滅菌製品のリリースの要求事項に関する情報
を提供している。
9.4.1.5
PQで使用する負荷は,日常的に滅菌する製品を代表するものでなければならず,日常処理する
負荷の中で最も滅菌困難なものに基づいて定義しなければならない。
9.4.1.6
載荷形態が大きく変わる施設(ヘルスケア施設)について,滅菌プロセスに影響を与える載荷形
態の変動の範囲を評価しなければならない。滅菌プロセスにばく露する全ての製品での要求するSALの達
成を立証しなければならない。
9.4.1.7
製品以外のものを用いる場合は,少なくとも滅菌プロセスに対して,製品より同等以上のチャレ
ンジでなければならない。
9.4.1.8
負荷をバリデーションサイクルに再度使用する場合は,作業員に対する労働安全規制に適合し,
負荷中のEO残留物が次の微生物学的PQ検討での微生物学的チャレンジに影響しないように,ばく露と
ばく露との間で適切にエアレーションしなければならない。
9.4.1.9
PQの一部にCIを使用する場合,これらはISO 11140-1に適合しなければならない。また,微生
物学的及び物理的監視と合わせて用いなければならない。
9.4.1.10 PQに用いるBIは,ISO 11138-1:2006の該当する箇条及びISO 11138-2:2006の箇条5及び9.5に
適合しなければならない。
9.4.2
稼働性能適格性の確認−微生物学的(MPQ)
9.4.2.1
微生物学的なPQ(MPQ)は,滅菌プロセスの適用において,あらかじめ定めた無菌性に対する
要求事項を満足することを立証しなければならない。この立証には,製造用チャンバを用い,あらかじめ
定めた滅菌プロセスより低い致死性を与えるように定めたプロセスパラメータを適用して実施しなければ
ならない。
9.4.2.2
MPQでは,製造用チャンバの中の製品/負荷の組合せについて,決定したプロセスの有効性を
確認しなければならない。
9.4.2.3
サイクルの致死率は,附属書A若しくは附属書Bに規定した方法,又は製品に要求するSALの
達成を立証する他の方法のうちの一つを用いて決定しなければならない。
9.4.2.4
研究用チャンバでプロセスを決定した場合,MPQでは,研究用チャンバで得られたデータを確
認できる製造用チャンバで少なくとも3回の部分サイクル又は3回のハーフサイクルを含まなければなら
ない。
18
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9.4.2.5
ハーフサイクル法によるオーバーキル法[B.1.2 a) 参照]を用いる場合,ハーフサイクル運転の
内部PCDは陽性があってはならない。
外部PCDが日常滅菌プロセスのための“ワーストケースチャレンジ”である内部PCDよりも大きな抵
抗性が立証されている場合,ハーフサイクルを実施しているときの外部PCDでのBIの陽性は容認できる。
しかし,全ての内部PCDは陰性であるのがよい。
9.4.2.6
サイクル計算法によるオーバーキル法[B.1.2 b) 参照]又はBI/バイオバーデン法(附属書A参
照)を用いる場合,幾つかの内部PCDが生残するかもしれないが,計算したSALはあらかじめ定めた値
に適合しなければならない(ISO 14161参照)。
9.4.3
稼働性能適格性の確認−物理的(PPQ)
9.4.3.1
物理的PQ(PPQ)では,次の両者を立証しなければならない。
a) 予定している日常のプロセス仕様で実行中に,負荷全体があらかじめ定めた合格基準に適合する。
b) プロセスの再現性。
PPQは計画した同一の検討において,全てのあらかじめ定めた合格基準に適合する連続で最低3回の適
格性の確認のサイクルを含まなければならない。PPQは,MPQの間に実施してもよい。PPQをMPQの少
なくとも3回のMPQと並行して実施する場合,最低1回の追加のPPQを全ての日常プロセス仕様で実施
しなければならない。
ある失敗の要因がバリデートしたプロセスの有効性に関連しない場合は,更に要求される3回連続した
PPQを行わないで,そのプロセスの実施と無関係であると文書化してもよい。これらの例として,停電,
その他のサービスの停止,外部監視装置の故障などがあるがこれに限定しない。
9.4.3.2
PPQでは,プロセスを次のように確認しなければならない。
a) 滅菌プロセスに入れる製品の最低温度及び/又はその温度を達成するのに必要な定義した条件を確立
しなければならない。
b) 定義したプレコンディショニング(行う場合)の終了時点で,滅菌負荷の温度及び湿度を確立してい
る。
c) プレコンディショニング(行う場合)の終了と滅菌サイクルの開始との間の定義した最大経過時間が
適切である。
d) 定義したコンディショニング(行う場合)の終了時点で,滅菌負荷の温度及び湿度を確立している。
e) パラメトリックリリースを適用する場合,チャンバの湿度が記録されている[9.5.5 a) 参照]。
f)
ガス状のEOが滅菌チャンバに導入されている。
g) 圧力の上昇及び使用したEOの量,又は滅菌チャンバ内のEO濃度を確立している[9.5.4 f) 参照]。
パラメトリックリリースを適用する場合は,9.5.5 b) も参照。
h) 滅菌サイクルの間,チャンバの温度,湿度(記録する場合)及び該当する場合は,その他のパラメー
タを確立している。
i)
ばく露中,製品負荷の温度を確立している。
j)
エアレーション(行う場合)の間,滅菌負荷の温度を確立している。
9.5
バリデーションのレビュー及び承認
9.5.1
この細分箇条の目的は,滅菌プロセス及び承認された手順/計画書によって合否を確認するために
バリデーションデータのレビューを行い,文書に記録し,プロセス仕様を承認することである。
9.5.2
BI培養試験結果を含めて,製品の決定,プロセスの決定,IQ,OQ及びPQにおいて収集し,作成
した情報は記録し,合否判定のためレビューしなければならない(4.1.2参照)。このレビューの結果は,
19
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
記録しなければならない。
9.5.3
バリデーション報告書を作成しなければならない。この報告書は,指定された責任者がレビュー及
び承認しなければならない。
9.5.4
バリデーション報告書は,適格とした製品,定義した載荷形態及びEO滅菌プロセスに関わる文書
化した仕様を記載又は参照し,次の事項を示さなければならない。
注記 実用的には,速度は,あらかじめ定めた圧力に到達するのに要した時間(許容幅を含む。)で求
めることができる。
a) 滅菌プロセスに入れる製品の最低温度及び/又は最低温度を達成するために必要な定義した条件。
b) プレコンディショニング(行う場合)
1) チャンバ内/エリアでの時間,温度及び湿度
2) 滅菌負荷の温度及び湿度
3) プレコンディショニングからの負荷の取出しから滅菌サイクルの開始までの最大経過時間
c) 真空度及び排気速度(行う場合)
1) 減圧保持時間(行う場合)
注記 排気速度は,一般的に,各運転の規定時間よりむしろ最小許容排気時間,最大許容排気時間又
は許容排気時間の範囲で規定される。
d) 不活性ガスによるフラッシング(行う場合)
1) 不活性ガス/水蒸気と合算される圧力(ΔP又は最終圧力)及び圧力到達速度(ΔP/時間)
2) 真空度(ΔP又は最終圧力)及び真空到達速度(ΔP/時間)
3) 繰返しの回数及び連続する繰返しでの全ての変動
e) コンディショニング及び/又は加湿保持フェーズ(行う場合)
1) 圧力水準及び/又は真空到達速度又は相対湿度水準(制御又は監視のいずれでも)
2) 水蒸気パルス/減圧(行う場合)の回数
3) 時間
4) チャンバ温度
5) コンディショニング終了時の滅菌負荷の温度及び湿度
f)
EO導入及びばく露
1) EO導入フェーズでのEO導入時の圧力上昇(ΔP),EO導入時間及び最終圧力
2) 圧力上昇及び次のいずれかによるガス状のEOが滅菌器チャンバ内へ導入されたことの証拠
i)
使用したEOの質量[D.10.2 i) 参照]
ii) EO濃度の直接測定
iii) 使用したEOの体積
3) 滅菌チャンバ温度
4) ばく露時間
5) 滅菌負荷の温度
6) ばく露中におけるチャンバ内ガス循環システム(行う場合)の正常な運転をしていることの表示
g) ばく露後のフラッシング(行う場合)
1) 真空度(ΔP又は最終圧力)及び真空到達速度(ΔP/時間)
2) 不活性ガス/水蒸気と合算される圧力(ΔP又は最終圧力)及び圧力到達速度(ΔP/時間)
3) 繰返しの回数及び連続する繰返しでの全ての変動
20
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
h) エアレーション(行う場合)
1) チャンバ及び/又は室内の温度及び湿度
2) チャンバ及び/又は室内の圧力変化(ある場合)
3) 空気,その他のガスの換気速度
4) 滅菌負荷の温度
9.5.5
パラメトリックリリースを実施する場合,バリデーション報告書には,次の事項を記載しなければ
ならない。
a) コンディショニング中のチャンバ内の湿度の直接測定による値及び許容範囲
b) 日常プロセスのプロセス仕様を確立するための分析方法を使用してチャンバ内雰囲気の直接分析から
得られたエチレンオキサイド濃度の値及び許容範囲。サンプリングはEOばく露の全体にわたって要
求される条件を検証するのに十分であるよう定められた間隔でなければならない。
c) チャンバの温度:2か所の別々の監視場所での記録
9.5.6
バリデーション中に得られた記録に基づいて,日常プロセスのためのプロセスパラメータ及びそれ
らの許容範囲を含んだプロセス仕様を確立しなければならない。このプロセス仕様には,指定したEOプ
ロセスを通った製品が適合した製品であり,リリースのための承認を得ることを示す判定基準も含めなけ
ればならない。
10
日常監視及び管理
10.1
日常監視及び管理の目的は,バリデートし,定められた滅菌プロセスがその製品に適用されている
ことを立証することである。
10.2
それぞれの滅菌サイクルについて,滅菌プロセスの仕様に適合していたことを証明するために,デ
ータを記録し,保管しなければならない。これらのデータは,少なくとも次の事項を含まなければならな
い。
注記 実用的には,速度は,あらかじめ定めた圧力を達成するために要した時間(許容幅を含む。)で
決定できる。
a) 滅菌プロセスに入れる製品の最低温度及び/又は負荷をじゅん(馴)化するために用いる定められた
条件
b) あらかじめ定めた場所で監視及び記録したプレコンディショニングエリア(行う場合)の温度及び湿
度
c) 各滅菌負荷のプレコンディショニング(行う場合)の開始及び負荷の取出し時刻
d) プレコンディショニング(行う場合)において滅菌負荷の取出しから滅菌サイクルの開始までの経過
時間
e) コンディショニング実施中及び/又は圧力,圧力上昇(ΔP)及び/又は直接測定による湿度保持フェ
ーズのチャンバ湿度
f)
コンディショニング時間
g) EO導入及びばく露中のチャンバ内ガス循環システム(行う場合)が正常な運転をしていることの表
示
h) 滅菌サイクル中のチャンバ内の温度及び圧力
i)
圧力を制御手段として用いる場合,少なくとも次の一つによって二次的な測定を行いEOのチャンバ
への導入を確認する。
21
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1) 使用したEOの質量[D.10.2 i) 参照]
2) 滅菌チャンバ内のEO濃度の直接測定
3) 使用したEOの体積
j)
EO導入時間
k) 不活性ガス導入(行う場合)
l)
ばく露時間
m) チャンバの排気にかかる時間
n) ばく露後のフラッシング中の時間及び圧力変化
o) エアレーション中の時間,温度及び圧力変化(ある場合は全て)
10.3
日常監視でBIを用いる場合は,8.6及び8.7に適合しなければならない。
日常的なリリースのために用いるPCDが,MPQに用いるものと異なる場合,MPQに用いるPCDとそ
のプロセスに対して同等以上の抵抗性であるとよい。
10.4
日常監視でCIを用いる場合は,8.8に適合しなければならない。
CIは,製品のリリースのためのBIを代替としてはならない。また,負荷をパラメトリックにリリース
する補助的な根拠に使用してはならない。
10.5
パラメトリックリリースを実施する場合は,次の追加データを記録し,保管しなければならない。
a) 滅菌サイクルを通して最低2か所のチャンバ内の温度。
b) コンディショニング中の直接測定したチャンバ内の湿度。
c) ばく露中を通して,要求される状態を検証するのに十分なあらかじめ定めた間隔で,分析的手法によ
るチャンバ雰囲気の直接分析から決定されるEO濃度
11
滅菌からの製品のリリース
11.1
それぞれの滅菌負荷に使用した滅菌プロセスの適合性を判断する基準は,文書化しなければならな
い。この基準は,次の事項を含まなければならない。
a) 日常プロセスで記録されたデータが,滅菌プロセス仕様に合致していることの確認。
b) 全てのBI(用いる場合)の試験微生物の生育を認めないことの確認。
注記 滅菌から正式に負荷をリリースするには,製品が流通網に入る前に,他の試験が必要になるこ
とがある(例えば,EO残留物,エンドトキシン,物理的試験など)。
11.2
プロセスが上記の全ての適合基準を満さない場合,その原因を調査しなければならない。装置の修
理又は変更が必要となる場合,このプロセスの使用を再開する前に必要な適格性の確認を実施しなければ
ならない。
11.3
11.1の適合基準に一つでも適合しない場合,製品は不適合であるとみなし,JIS Q 13485の該当す
る箇条に従って取り扱わなければならない。BIが陽性となった場合,製品の無菌試験が許容できる結果に
なっても製品のリリースはできない。
不適合は文書化した手順に従い処理しなければならない。
11.4
バリデーション検討を販売できる製品で行った場合は,流通させる製品のリリースのための要求事
項をバリデーション活動の開始の前に作成しなければならない。製品のリリースの前に,バリデーション
/滅菌プロセスへの繰返しのばく露の製品及び包装機能への影響及びEO残留物及び/又は反応生成物を
評価することが重要である。
MPQを販売できる製品で行った場合,市場にリリースする前に,製品がフルのばく露の滅菌プロセスに
22
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
かけ,その受入れについて正式なレビューにかけたことを確実にする手順を確立しなければならない。
注記 単一ロットのリリースについての情報は,附属書Eを参照。
12
プロセス有効性の維持
12.1
一般
12.1.1
滅菌プロセスに提供する製品の状態を確実にするために,システムの継続的な有効性を立証しな
ければならない(7.3.1参照)。
12.1.2
滅菌プロセスの制御及び監視に使用する計器の正確度及び信頼性は,4.3.3に従って定期的に検証
しなければならない。
12.2
装置のメンテナンス
12.2.1
予防メンテナンスは,文書化した手順で計画し実施しなければならない。全ての手順は,関連す
る国又は地方の要求事項とともに滅菌器の製造業者の推奨事項によらなければならない。
12.2.2
あらかじめ定めた全てのメンテナンス業務を完遂し記録するまで,装置は製品の滅菌処理に使用
してはならない。
12.2.3
メンテナンスの記録は,保管しなければならない(4.1.2参照)。
12.2.4
メンテナンスの計画,メンテナンスの手順及びメンテナンスの記録はあらかじめ指名した職員が
定めた実施頻度でレビューし,その結果は文書化しなければならない。
12.3
適格性の再確認
12.3.1
必要とする適格性の再確認の範囲を決定するために,IQ,OQ,PQとその後に実施した適格性の
再確認を年1回レビューしなければならない。これには微生物学的検討でのSALの再確認の必要性の評価
を含めなければならない。このレビューの結果は,得られた理由とともに文書化しなければならない。
12.3.2
あらかじめ定めた装置で実施する滅菌プロセスの適格性の再確認は,適合基準及び文書化した手
順によって,定めた間隔で実行しなければならない。この間隔は,正当な理由付けをしなければならない。
12.3.3
適格性の再確認によってその滅菌プロセスが要求される製品のSALをもはや達成できない場合,
その不具合の原因を究明し是正及び/又は予防処置をとらなければならない。この検討の一部として,既
に処理した製品の負荷について規定したSAL達成への影響を考慮し,製品の使用の適切さについてのリス
ク評価を実施しなければならない。この検討の結果,要求されるSALをもはや達成できないことが判明し
た場合,要求されるSALの再確立のために,新たなMPQ及びPPQを実施しなければならない。この検討
及びその後の処置は記録しなければならない。
12.3.4
適格性の再確認データ,報告書,及びその結果で実施した是正処置(必要な場合)のレビューの
記録は,保管しなければならない(4.1.2参照)。
12.4
変更の評価
12.4.1
製造方法,製品,滅菌器及び/又は滅菌プロセスの変更は,滅菌プロセスの有効性への影響につ
いて評価しなければならない。
12.4.2
製品のバイオバーデンに関連した内部及び/又は外部PCDの妥当性は,変更後でも適切であるか
を再確認しなければならない(8.6及び10.3参照)。
12.4.3
負荷及び載荷形態は,変更後,その適切性を再評価しなければならない。そしてこの再評価の結
果は,4.1.2に従い文書化(記録)しなければならない。
12.4.4
適格性の確認を実施した滅菌プロセスの有効性に変化を与える可能性のある変更を滅菌プロセ
ス,滅菌器又は製品に加えたときにはいつでも滅菌プロセスをレビューしなければならない(8.2参照)。
23
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
12.4.5
変更の大きさを考慮してプロセスの決定,IQ,OQ又はPQの実施範囲を決定しなければならない。
12.4.6
この評価の結果は決定に至った理由を含めて,文書化しなければならない。
12.5
同等性の評価
12.5.1
プロセスの同等性
IQ及びOQを実施済みの,同一のプロセスパラメータを提供する滅菌器は,次のいずれかによってその
適格性を確認しなければならない。
a) 元の滅菌器と同一の方法
b) 要求するレベルの微生物学的致死率が得られる規模を削減したMPQ,並びに製造用チャンバ内での負
荷の温度・湿度の均一性及び制御を立証するPPQ。この規模を削減した適格性の確認の根拠は記録し
文書化しなければならない。
製品及び負荷の特性に対する地理的に異なった場所による影響は評価しなければならない。
12.5.2
製品
ある製品が適格性の確認を実施済みの既存の製品又は内部PCDより同等以下の抵抗性をもつとみなせ
る場合,バリデートしたプロセスに加えてもよい。既存のEOプロセスをバリデートするために使用した
製品及びPCDと候補の製品とを比較し,技術的なレビューをしなければならない。この技術的なレビュー
の結果は,この製品を受け入れることの決定についての根拠を含めて文書化しなければならない。7.2の要
求事項はこの製品についても適用する必要がある。
24
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書A
(規定)
滅菌プロセスの致死率の決定−
バイオロジカルインジケータ/バイオバーデン法
A.1 一般
A.1.1 この方法は,滅菌プロセスパラメータ(滅菌サイクルばく露時間)を確立するための,あるプロセ
スでのBIの抵抗性の知見と,バイオバーデン数及び抵抗性の知見とを組み合わせた方法である。
この方法の採用は,製品のバイオバーデンレベルが長い間比較的一定で,バイオバーデンの抵抗性はBI
の抵抗性と同じか又は低いことの立証を要求する(D.8.6参照)。
内部PCDの抵抗性は,滅菌サイクルにばく露するとき,そのプロセスを段階的なばく露時間又は段階的
な菌数のBIを単一の滅菌ばく露時間で運転し,そのプロセスの致死率(D値の計算による不活化速度)を
測定して立証する。このBIの死滅速度,バイオバーデンの菌数及び相対的抵抗性の知見によってSALを
予想できるばく露時間を設定することができる。
包装の影響及びPCDからのEOの除去に注意を払わなければならない。
この方法の指針についてはISO 14161を参照。
A.1.2 培養時間を含めて,適格性の確認に使用するBIの回収に用いる条件を確立し文書化しなければな
らない。培養時間は,EOにばく露した芽胞の生育の遅延の可能性があることを考慮しなければならない。
BIの培養時間についての追加の情報はISO 14161を参照。
A.1.3 ほかの全てのパラメータは同じままで,段階的に設定した時間でEOにばく露した後,又は段階的
な菌数のBIへのEOばく露をした後のプロセスの致死率は,次のいずれかの方法で決定できる。
a) 直接計数法
b) フラクションネガティブ法
c) 上記のa) 及びb) の組合せ
注記 フラクションネガティブ法は,標準菌を用いて部分的なガスばく露時間でばく露した後,又は
段階的に設定した標準菌の菌数を1回のガスばく露時間にかけた後の培養試験(無菌性の試験)
での陽性/陰性データを用いる。
A.2 手順
プロセス開発について追加の指針は,AAMI TIR16及びISO 14161を参照。両者はプロセス開発につい
て詳細に述べている。
25
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書B
(規定)
安全率を見込んだ滅菌プロセスの致死率の決定−
オーバーキル法
B.1
一般
B.1.1 プロセスを決定するこの方法は,標準菌の不活化に基づいており,広く採用されている(ISO
11138-2参照)。この方法で実施される滅菌プロセスは,安全率を見込んだ方法であることが多く,無菌性
に関してあらかじめ定めた要求事項の達成よりも必要以上の処理をしてしまうことがある。
この方法の指針は,ISO 14161参照。
B.1.2 このプロセスを決定する方法には,次のa) 又はb) のいずれかの方法を用いることが必要である。
a) ハーフサイクル法:最低ばく露時間を確認するために,BI(106以上の菌数で,適用する場合はPCD
に入れて)の全てが不活化の結果となるような計3回の連続した実験を実施しなければならない。定
めるばく露時間は,少なくともこの最低時間の2倍でなければならない。BIの生残が確認できる短い
サイクルも,EOガスにばく露したBIの回収技術が十分に適切であることを証明するために実施しな
ければならない。
注記 この短いサイクルはBI,PCD及び製品のバイオバーデンの相対的な抵抗性を示すのにも使用
できる。
b) サイクル計算法:A.1.3に規定する方法の一つを用いて,BIの12 SLRを最低限度与える日常プロセス
パラメータを確立しなければならない。サイクルの回数は,用いた方法によって決まる。
B.1.3 適格性の確認の検討で使用したBIの回収のための条件は確立し,文書化しなければならない。培
養時間は,EOにばく露した芽胞の生育の遅延の可能性があることを考慮しなければならない。BIの培養
時間についての詳細な情報はISO 14161を参照。
B.1.4 製品のバイオバーデンの抵抗性は,製品のバイオバーデンの全不活化時間が製品のBI(内部PCD)
よりも短いことなどで示さなければならない。
B.2
手順
B.2.1 滅菌条件の達成が最も困難な場所の製品内にBIを置く又は接種することによってEOに対して既
知の抵抗性及び既知の数の微生物を含めた滅菌プロセスへのチャレンジ(PCD)を作製する。微生物学的
チャレンジの設置場所が製品中で最も滅菌が困難な場所以外である場合は,その場所と滅菌が最も困難な
場所との関係を確立しなければならない。
B.2.2 製品よりも滅菌プロセスに対して微生物学的な抵抗性が同等以上であることが示されるPCDの使
用は,この要求事項に適合する。包装の影響及びPCDからのEOの除去に注意を払う必要がある。
B.2.3 PCD(B.2.1及びB.2.2に従い)は,滅菌負荷の中又は上に適切に置く。
B.2.4 滅菌負荷を,あらかじめ定めた滅菌プロセスより低い致死性を与えるように設定した条件下でEO
にばく露する。
B.2.5 サイクル計算法として既知の微生物数の不活化をA.1.3によって確認した場合は,定めたSALに
対する微生物の生残確率を外挿法によって推定し滅菌プロセスの処理時間を決める。
26
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書C
(参考)
温度センサ,湿度センサ及びバイオロジカルインジケータの数
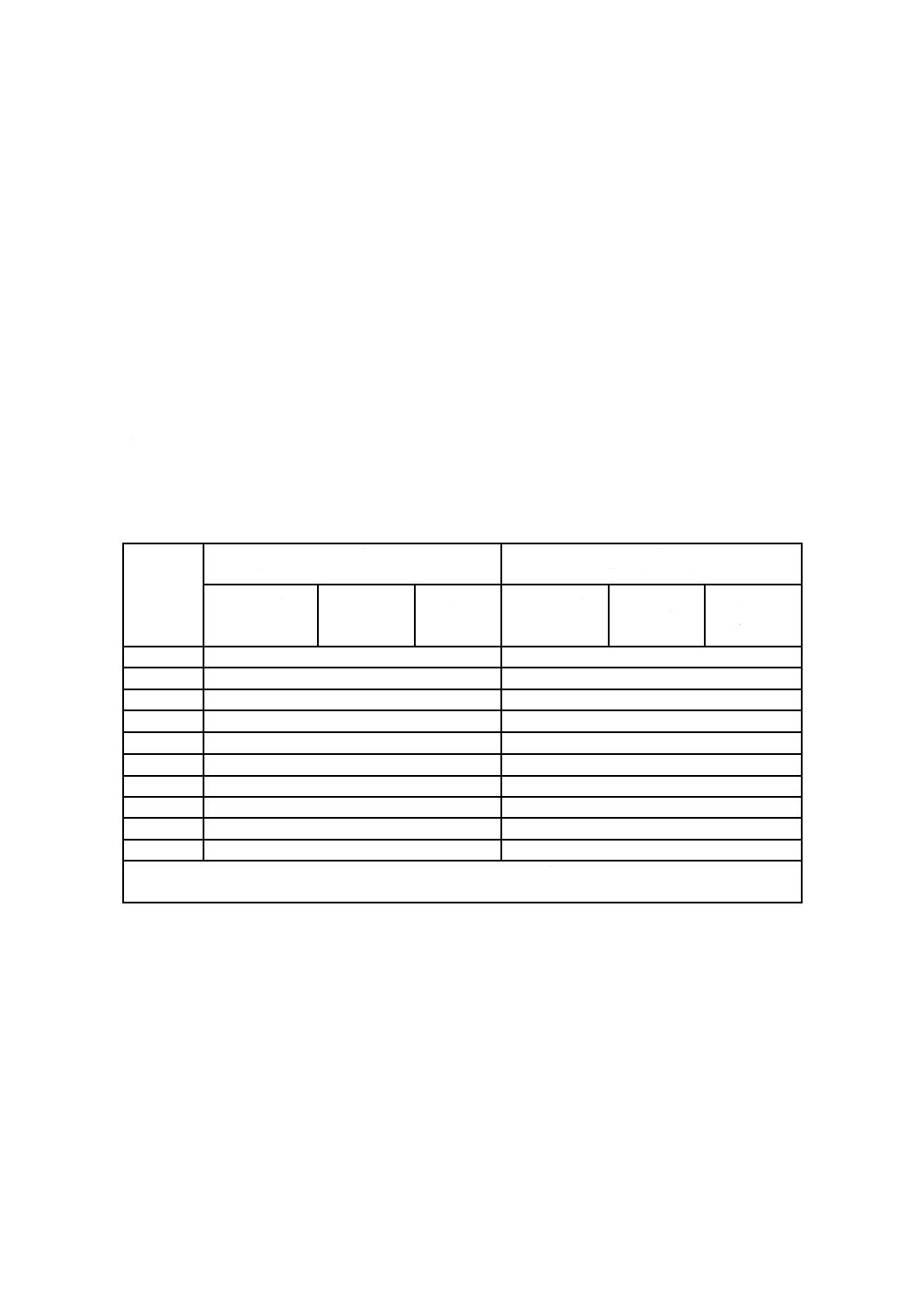
C.1 温度センサ
ホット,又はコールドの可能性のある位置の特定を目的として,部屋又はチャンバの温度分布図を作成
するために,OQの間,2.5 m3当たり1センサを用いることが望ましい。監視点は立体的に置き,かつ,扉
の近くの場所を含むとよい。
PQでは,製品容積の1 m3当たり1温度センサを必要とする。最低限の温度センサの数は3である。セ
ンサは(可能な場合)負荷内の包装の中(例 無菌バリアシステムの中,又は個別の包装単位の間)に置
くとよい。
計算の結果,端数は整数に切り上げる。
温度センサ数は,表C.1によるとよい。
表C.1−推奨する温度センサの最低数
容積
(m3)
OQの場合の数
(有効チャンバ容積/部屋の容積)
PQの場合の数
(製品負荷容積)
プレコンディシ
ョニング
コンディシ
ョニング
/滅菌
エアレーシ
ョン
プレコンディ
ショニング
コンディシ
ョニング
/滅菌
エアレーシ
ョン
≦1
3
3
10
4
10
15
6
15
20
8
20
25
10
25
30
12
30
35
14
35
40
16
40
50
20
50
100
40
100
例1 OQ時,プレコンディショニング室の有効チャンバ容積70 m3:70/2.5=28。
例2 PQ時,製品負荷容積2 m3:2/1=2。用いるセンサの数は,3個以上(使用するセンサの最低数)。
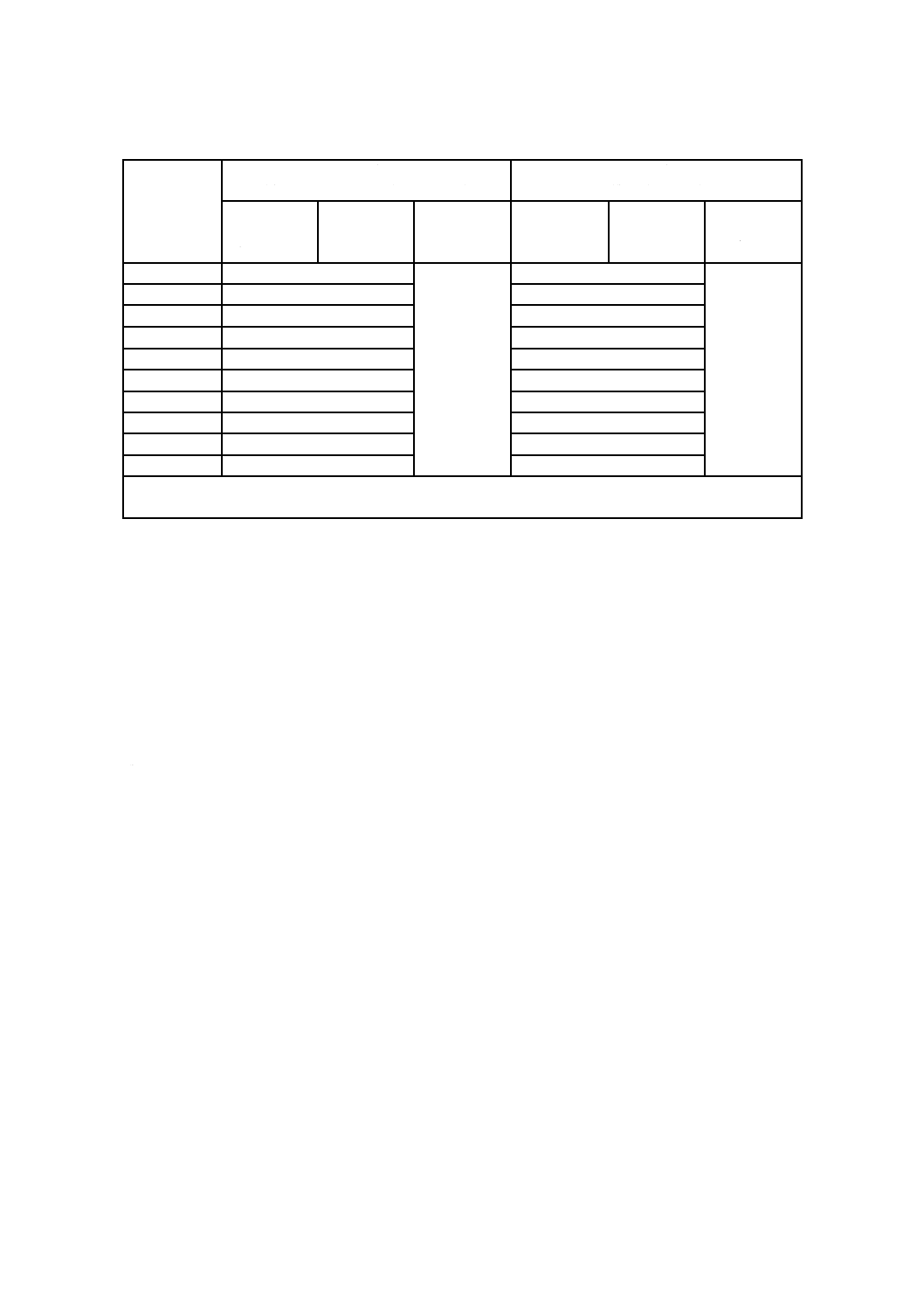
C.2 湿度センサ
湿度レベルの変動範囲の特定を目的として,そのエリア又は製品の湿度分布図を作成するために,2.5 m3
当たり1センサを用いることが望ましい。センサの最低数は2個である。
計算の結果,端数は整数に切り上げる。
PQでは,センサは(可能な場合)負荷内の包装の中(例 無菌バリアシステムの中,又は個別の包装
単位の間)に置くとよい。
湿度センサ数は,表C.2によるとよい。
27
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表C.2−推奨する湿度センサの最低数
容積
(m3)
OQの場合の数
(有効チャンバ容積/部屋の容積)
PQの場合の数
(製品負荷容積)
プレコンデ
ィショニン
グ
コンディシ
ョニング/
滅菌
エアレーシ
ョン
プレコンデ
ィショニン
グ
コンディシ
ョニング/
滅菌
エアレーシ
ョン
≦1
2
適用しない
2
適用しない
10
4
4
15
6
6
20
8
8
25
10
10
30
12
12
35
14
14
40
16
16
50
20
20
100
40
40
例1 OQ時,6 m3の有効チャンバ容積:6/2.5=2.4。用いるセンサの数は,3個以上である。
例2 PQ時,製品負荷容積60 m3:60/2.5=24。用いるセンサの数は,24個以上である。
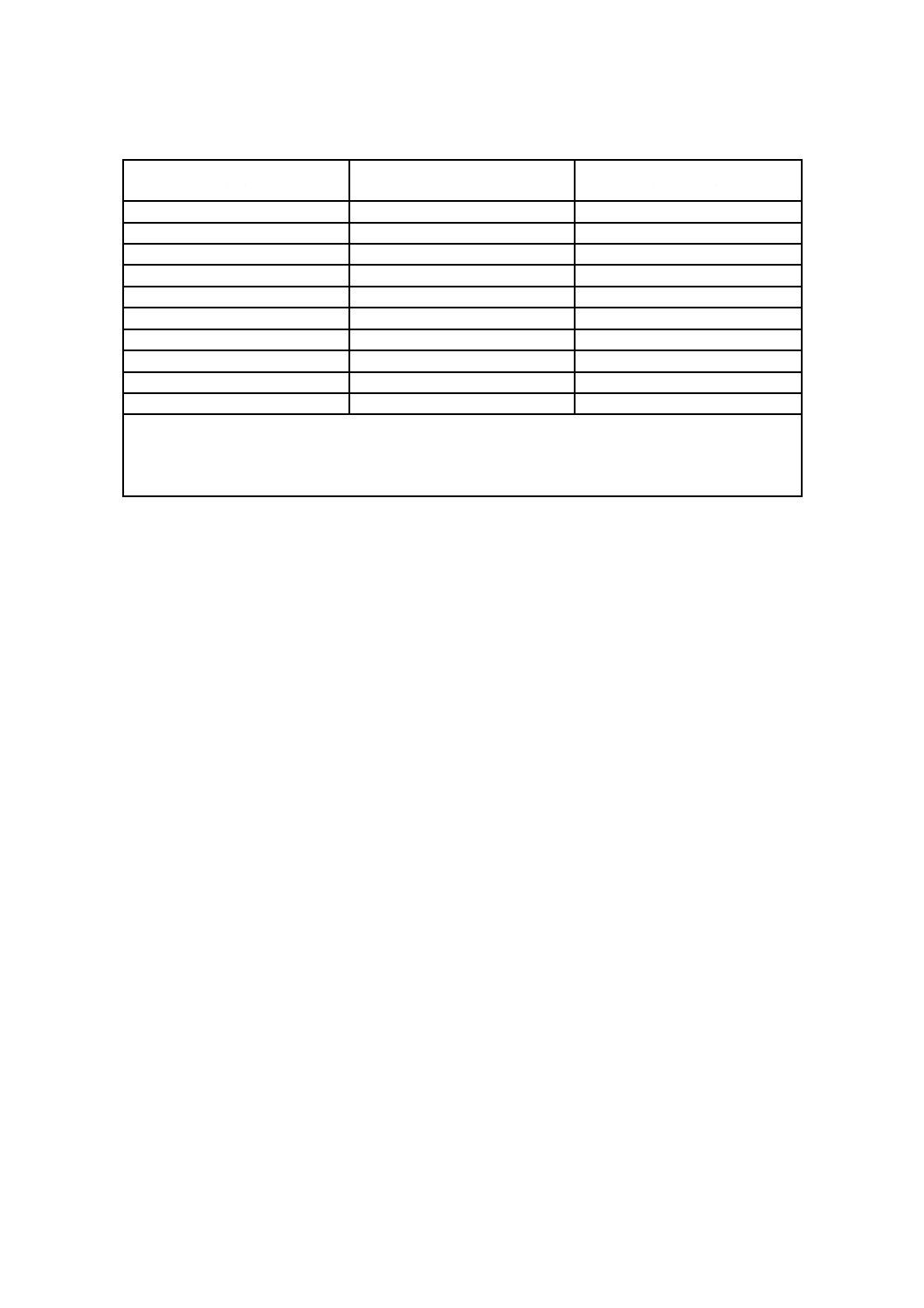
C.3 バイオロジカルインジケータ(BI)
推奨するBI/PCDの最低数は次による。
a) MPQについて,製品負荷容積10 m3まで,BIの数は製品容積m3当たり3個,最低5個。
b) MPQについて,製品負荷容積10 m3超え,1 m3当たり1個を追加する。
日常管理のためBIを用いる場合の数は,MPQで用いるBI数の半数で,最大は30個とする。
BI/PCD数は,表C.3によるとよい。
用いるBI/PCDの実際の数は,次による。
a) 選択した微生物学的方法(附属書A又は附属書Bを参照)
b) 製品容積
c) チャンバのタイプ(研究又は製造)
ストゥンボ・マーフィー・コクラン法及びオーバーキル法のサイクル計算法を用いているときの推奨さ
れるBI/PCDの数は,処理する製品容積を基礎とすることができる。この方法を用いるとき,最低10個の
BI/PCDを必要とする[38]。
28
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表C.3−推奨するBI/PCDの最低数の例
製品負荷容積
(m3)
MPQ
日常管理
(用いる場合)
≦1
5
3
10
30
15
15
35
18
20
40
20
25
45
23
30
50
25
35
55
28
40
60
30
50
70
30
100
120
30
例1 3 m3の製品負荷容積:3×3=9。用いるBIの数は,MPQでは9個以上である。
日常管理:9/2=4.5。BIの数は5個以上。
例2 18 m3の製品負荷容積:10×3+(18−10)×1=38。用いるBIの数は,MPQでは38個以上である。
日常管理:38/2=19。BIの数は19個以上。
29
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書D
(参考)
規定要求事項の適用に関する指針
この附属書に示す指針は,この規格に適合しているかを評価するためのチェックリストとして意図した
ものではない。この指針の意図するところは,規定された要求事項を達成するための解説及び可能な方法
を提示することによって,この規格の統一的な理解及び実施するための手助けとなることである。この規
格に適合することを立証することによって,この指針で示す以外の方法を適用することも可能である。
注記 参照を容易にするために,この附属書の箇条番号は,この規格の規定部分の箇条に対応してい
る。
D.1 適用範囲
指針はない。
D.2 引用規格
引用規格として含まれる文書に示される要求事項は,この規格の規定部分に記載されている範囲だけが
要求事項である。引用内容は規格全体又は特定な箇条に限定できるが,後者の場合,引用規格は西暦年を
付記している。
D.3 用語及び定義
指針はない。
D.4 品質マネジメントシステム(QMS)
注記 JIS Q 13485の適用範囲は医療機器の製造業者に焦点を当てているので,ヘルスケア施設はその
組織に適用できる他の品質マネジメント規格を用いることができる。
D.4.1 文書化
JIS Q 13485参照。
D.4.2 経営者の責任
D.4.2.1 責任及び権限に関わる要求事項は,JIS Q 13485:2005の5.5に,人的資源に関わる要求事項は,
JIS Q 13485:2005の6.2に規定されている。
JIS Q 13485:2005では,経営者の責任に関する要求事項は,経営者のコミットメント,顧客重視,品質
方針及び計画,並びに責任,権限,コミュニケーション及びマネジメントレビューに関連している。
各組織は,どのような訓練が必要かを識別/判断する方法を定め,全ての職員が割り当てられた責任を
十分に果たすことができるような訓練を確実に行うとよい。
D.4.2.2 滅菌プロセスの開発,バリデーション及び日常管理は,幾つかの別々の組織を包含することがあ
り,それぞれがある要素に責任をもつことがある。この規格の要求事項に合致するために,それぞれの組
織での手順で,責任範囲を明確にすることが重要である。これは受託者が業務の実施を受託した場合は特
に重要である。
滅菌プロセスの要素を外部委託するとしても,医療機器製造業者は最終的にバリデーション,出荷及び
30
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
滅菌された製品の市場への流通に責任があることに注意することは重要である。ヘルスケア施設が再使用
可能医療機器の滅菌を外部委託する場合,バリデーション及び滅菌製品のリリースはヘルスケア施設の責
任である。
詳細な指針はISO 14937:2009のE.4.2.2参照。
D.4.3 製品実現
注記 JIS Q 13485では製品実現に対する要求事項は,顧客要求の決定,設計・開発,購買,製造管理,
監視及び測定機器の校正からなる製品ライフサイクルに関連している。
D.4.3.1 JIS Q 13485:2005の7.4において,購買に関わる要求事項が規定されている。特に,JIS Q
13485:2005の7.4の購買製品の検証に関わる要求事項は,外部の組織から受け取るプロセスの質に影響を
与える製品及びサービスに適用されることに注意するとよい。
ヘルスケア施設での購買手順には,再使用可能医療機器が,洗浄,消毒,滅菌及びエアレーションのた
めのISO 17664にあらかじめ定められたバリデートした指示書とともに提供されることを確実にするとよ
い。ヘルスケア施設でこの指示書に述べられた洗浄,消毒,滅菌,エアレーションのため手順が実施可能
であることも検証するとよい。
D.4.3.2 識別及びトレーサビリティに関わる要求事項は,JIS Q 13485:2005の7.5.3に規定されている。
JIS Q 13485に完全には適合していない施設,例えば,ヘルスケア施設については,製品の識別及びトレ
ーサビリティの維持の手順には,滅菌前の各品物又は包装のラベルを含めたロット管理とともに次の情報
を含めるとよい。
a) 滅菌器ID又はコード
b) 滅菌日
c) サイクル番号(サイクル実施日又は滅菌器のサイクル運転番号)
d) そのパックを組み立てた人の特定
問題が発生した場合,その包装を組み立てた人の特定を含めることによって,更に詳細な調査が可能で
ある。ロット識別情報によって,リコールの場合には特定のサイクルで滅菌された品物の回収が可能で,
それらの原因までトレースが可能となる。
D.4.3.3 監視及び測定機器の校正に関わる要求事項は,JIS Q 13485:2005の7.6に規定されている。
D.4.4 測定,分析及び改善−不適合製品の管理
不適合製品の管理及び是正処置の手順は,JIS Q 13485:2005の8.3及び8.5.2にそれぞれ規定されている。
D.5 滅菌剤の特性
D.5.1 一般
指針はない。
D.5.2 滅菌剤
EOは多くの包装材及び高分子材料に対する浸透性の高いガスである。よく知られている配合は純EO及
び二酸化炭素又は窒素との混合物である。
注記 二酸化炭素,窒素又は他の不活性ガスとEOガスを混合する場合,高分子材料へのEO分子拡
散速度は滅菌剤中のEOガスの容積パーセントが影響する。要求する微生物の芽胞対数減少の
達成には,結果として長いEOばく露時間となる。
EOの貯蔵条件及び有効期限は,EO製造業者の推奨事項及び全ての適用される規制に従うとよい。この
遵守は層分離が問題となる混合ガスについて特に重要である。
31
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
D.5.3 微生物殺滅効果の有効性
指針はない。
D.5.4 材料への影響
指針はない。
D.5.5 安全性及び環境
D.5.5.1 EOは毒性,可燃性及び爆発性がある。したがって,その取扱い及び使用に十分な注意が必要で
ある。爆発限界は空気中でEOの体積分率2.6 %〜100 %である。
実際的に,EO滅菌サイクルは,爆発のリスクを最小限度にするために滅菌サイクル全体を通して非可
燃域で操作するとよい。これはEOガスの導入前にチャンバからの空気の除去が要求される。100 % EO滅
菌プロセスでは高真空引き又は数回の部分真空引きの後に,例えば,窒素のような不活性ガスの導入によ
る置換によって空気除去が実施できる。チャンバからの空気の除去はチャンバへEOガスを安全に導入さ
せるためである。EOガスばく露段階の完了のとき,爆発限界2.6 %以下のガスのレベルまでチャンバから
EOガスを除去することが必要である。これは真空引き/窒素を導入の繰り返しで達成される。
非可燃性混合滅菌剤の使用は火災又は爆発のリスク減少によって安全性を改善できる。また,国の装置
安全性要求事項を容易に遵守できる。非可燃性混合滅菌剤は,可燃性の高いEOガスを一つ以上の不活性
ガスと混合して製造する。そのような混合物の可燃性は滅菌器内のEO,空気,希釈ガス(例えば,二酸
化炭素など),不活性ガス(例えば,窒素)及び水蒸気の相対比率の測定によって評価できる。安全性及び
品質の問題を起こすようなEO混合物の分離がないことの確認に注意を払うとよい。
EO滅菌器は専用の部屋に据え付けるとよい。滅菌装置の制御操作は,滅菌室に入ることなく,作業者
がプログラムパラメータを設定及び変更できるようにその部屋の外に装備するとよい。滅菌器のアクセス
区域からの空気の流れは全て外に排気し,適用される要求事項に適合するとよい。
滅菌器から製品を取り出す前に,製品からのEOの放出によって作業者が関連するばく露限界[許容ば
く露限界(PEL:permissible exposure limit)/短時間ばく露限界(STEL:short term exposure limit)]を超え
る高レベルのEOとなる負荷の放出ガスにばく露されないように予防策をとるとよい。EO・不活性ガス混
合物で滅菌した製品をサイクルの終了時に滅菌器からすぐには取り出さない場合,滅菌器の中のEO濃度
は人への安全性についての問題を引起こす場合がある。
D.5.5.2 環境マネジメントシステムの原則は,EO滅菌プロセスに適用できる。JIS Q 14001は,環境マネ
ジメントシステムの仕様を提供している。JIS Q 14040は,ライフサイクルアセスメントの設計指針を提供
している。
D.5.5.3 排出ガスは,酸化触媒,湿式酸性スクラバー(気体洗浄機)又は燃焼などによるEOガス処理シ
ステムを通して,地方の許容基準又は排出管理規制に適合するように排出するとよい。
希釈剤の選定において,全ての副生成物の廃棄と同様に希釈剤のオゾン破壊の可能性についても考慮す
るとよい。
D.6 プロセス及び装置の特性
ヘルスケア施設において,プロセス及び装置の特性付けは一般的には滅菌器製造業者の責任である。ヘ
ルスケア施設のマネジメント(経営者)は,購入する装置が国及び地方の規制に適合していること,及び
その装置がEO滅菌をしようとする製品に対して適切かつ確実とするため,適切な管理をするとよい。ヘ
ルスケア施設のマネジメントは,滅菌する装置の正常な運転,及び医療機器の効果的な滅菌の達成に必要
なインフラストラクチャーをその施設が確実に装備するとよい。
32
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
D.6.1 一般
指針はない。
D.6.2 プロセスの特性
D.6.2.1 指針はない。
D.6.2.2 EOによる微生物の不活化に対する抵抗性は含有する湿気に影響される。30 %以下の低湿度では,
湿度の減少に従い,一部の製品では微生物学的抵抗性が増加する場合がある。そのため,微生物の含有す
る湿気とその周囲の湿度を平衡化させることを目的として,一般的に,製品がばく露される雰囲気中の湿
度を制御及び監視する。包装した製品について,過剰に高い相対湿度が製品の機能及び包装の完全性に影
響を与えないことを確実にするための考慮をするとよい。製品内への湿度到達を補助する一つの方法が,
定義した温度及び湿度での製品のプレコンディショニングである。そのようなプレコンディショニングは
滅菌サイクルの時間を短縮できる。ヘルスケア施設では,洗浄後の不十分な乾燥によって,製品が過剰に
高い湿気を含む可能性がある。
製品の加温及び加湿は,EOばく露前に,製品の再現性ある温度及び湿度を確立するために行う。プレ
コンディショニングのセル/部屋に置かれる最低時間の設定実験によって,滅菌負荷において要求される
条件の達成を確実にする。滅菌負荷での過剰な結露を防ぐために予防策をとるとよい。
プレコンディショニングを別のチャンバ,部屋又はセル内で実施するのは一般的であるが,滅菌チャン
バ内においてコンディショニングフェーズ中に,要求される負荷内での温度及び湿度の範囲を達成するよ
うに滅菌サイクルを設計することも可能である。滅菌プロセスのプレコンディショニング及びコンディシ
ョニングフェーズ中は,過剰な結露のリスクを最小にするために,負荷の温度をプロセスの環境の露点温
度以上に保持することを推奨する。
プレコンディショニングの終了時の滅菌負荷内の実際の温度及び湿度の範囲はPQのときに立証すると
よい。
該当する場合,プレコンディショニングから負荷を取り出して滅菌サイクルを開始するまでの間の最大
時間を確立する必要がある。60分以下の移送時間が一般的である。
a) プレコンディショニングなしに滅菌チャンバへ製品を入れる場合,製品及び包装内の過剰な結露の可
能性を考慮するとよい。
b) EO及びその反応物の残留は有害となる可能性がある。滅菌製品の製造業者は,製品に残留物が存在
する可能性を知っておかなければならない。エアレーションの全ての効率に影響する温度,保持時間,
強制加温空気循環,負荷特性,製品及び包装材料,並びに設定値及び許容値は,JIS T 0993-7に概説
されている残留量の評価のときに考慮するとよい。エアレーションは滅菌器内,分離したエリア,又
は両者の併用で実施できる。ヘルスケア施設では,EOにばく露される危険を避けるために,普通は
エアレーション用の部屋内よりむしろ滅菌器のチャンバ内でエアレーションが実施される。ヘルスケ
ア施設では,EO滅菌で再生処理した品物は医療機器及び滅菌コンテナの製造業者の推奨事項に従っ
て,取扱い又は使用する前に入念にエアレーションする必要がある。エアレーションが不十分な品物
及び包装はEOを放出し,患者及びヘルスケア施設の職員に危害を与える可能性がある。
D.6.2.3 移送時間とは,プレコンディショニングから取り出し,サイクルを開始するために製品を滅菌器
内へ搬入する間の時間を指す。
D.6.2.4 次の項目は,滅菌サイクルに含めることができるフェーズとそれらの各フェーズで考慮するとよ
い性能ファクターのリストである。
a) 空気の除去
33
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1) 到達真空度(ΔP又は最終圧力)及び真空到達速度(ΔP/時間)
b) 該当する場合,チャンバリーク試験[真空サイクルについては,減圧下(大気圧以下)で実施するか,
又は加圧サイクルについては加圧下(大気圧以上)及び減圧下(大気圧以下)で実施する。]
1) 安定時間及び/又は保持時間
2) 圧力変化
c) 不活性ガスの追加(行う場合)
1) 不活性ガスの導入による,圧力(ΔP又は最終圧力)及び圧力到達速度(ΔP/時間)
d) コンディショニング(行う場合)
1) コンディショニング中の水蒸気の導入による圧力上昇(ΔP又は最終圧力)又は相対湿度(%),及
び圧力到達速度(ΔP/時間)
2) 該当する場合,水蒸気パルス/真空引きの回数
e) EO導入
1) EOの導入によるあらかじめ定めた圧力に到達するときの圧力,圧力上昇(ΔP)及び速度(ΔP/時
間),並びにEO濃度監視に用いた方法との相関
2) 全ての不活性ガスの導入(行う場合)による,あらかじめ定めた圧力に到達するときの圧力,圧力
上昇(ΔP)及び速度(ΔP/時間)
f)
ばく露時間のあらかじめ定めた条件の維持
1) 滅菌剤又は不活性ガスを用いた圧力調整(行う場合)
2) チャンバ温度
g) EO除去
1) EOを除去するための到達真空度(ΔP又は最終圧力)及び真空到達速度(ΔP/時間)
h) フラッシング(行う場合)
1) 圧力上昇及び圧力到達速度
2) EOを除去するための到達真空度(ΔP又は最終圧力)及び真空到達速度(ΔP/時間)
3) 繰返しの回数及び連続した繰返しにおけるバリエーション
i)
空気/不活性ガスの導入
1) 不活性ガス又は空気導入による到達圧力(ΔP又は最終圧力)及び圧力到達速度(ΔP/時間)
2) 繰返しの回数及び連続した繰返しにおけるバリエーション
3) 空気導入による大気圧への復圧
D.6.2.5 製品の残留レベルを評価する際,循環速度をあらかじめ定めておくとよい。
D.6.3 装置の特性
D.6.3.1 次のファクターは装置を特性付けるときに考慮するとよい。
a) プレコンディショニングエリアの特性
プレコンディショニングは別に設けたプレコンディショニングエリア(チャンバ,セル又は部屋)
で実施できる。エアロゾル噴霧器(例えば,回転円盤加湿器及びネブライザー)のような加温されな
い水の散布によって動作する加湿器は微生物の潜在的な汚染源となるので,水蒸気による加湿が必要
である。
プレコンディショニングエリア(用いる場合)は,次の性能及び監視機能があるとよい。
− 有効空間の温度及び湿度の均一性を確実にするため,及び負荷を入れた部屋又はチャンバ内の温
度と湿度との均一性が維持されることを確実にするための,十分な空気循環。
34
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
− 前もって決定した許容範囲内であることを確実にする,気流感知装置,警報システム又は循環シ
ステムの監視表示
− プレコンディショニングエリアへの負荷の搬入及び搬出の時刻を記録する方法
− セル/部屋の温度及び湿度の監視手段
− セル/部屋の温度及び湿度の制御手段
b) 滅菌器の特性
滅菌器チャンバは次の性能及び監視機能があるとよい。
− 時間,チャンバ圧力,温度及び湿度(湿気の添加がセンサの読みで制御されている場合)を監視
する方法
− 時間,チャンバ圧力,温度を制御する方法及び湿気の添加がセンサの読みで制御されている場合
は湿度を制御する方法(装置に設置されているセンサの場合,IQ又はOQの間に圧力上昇との関
係を確実にする。)
− 湿度がセンサの測定値で制御されていない場合は,水蒸気添加を監視及び制御する方法
− パラメトリックリリースを用いる場合,コンディショニング中の湿度,及びEOばく露時間中の
EO濃度の直接分析のための分析装置(9.5.5及びD.9.5.5参照)
− チャンバへガス状のEOの導入を制御するシステム
− チャンバへガス状のEOが導入されたことを立証する方法。これは気化器から滅菌チャンバへ流
れるEOガスの温度の測定によってできる。このシステムによって,EOばく露時間中のEO濃度
を管理可能である。
− 適宜適切な処置を行うことができるようにサイクルパラメータの逸脱を検出及び警告する方法
c) エアレーションエリアの特性
エアレーションエリア(チャンバ,セル又は部屋)は製品/包装からEO残留物を除去するために
用いることができる。そのエリアの全体での温度均一性,新鮮空気の追加及び再循環空気は一定及び
再現性のある結果を確実に得るために重要である。エアレーションエリアには次の性能及び監視機能
があるとよい。
− 負荷を入れた部屋又はチャンバ内の十分な気流が前もって決定した許容範囲内で運転及び維持さ
れていることを確実にする気流感知装置,警報システム又は空気処理システムの監視表示器
− 空気を再循環する装置
− 室温を監視する方法
− 室温を制御する方法
D.6.3.2 法規及び安全性の要求事項に適合し,技術仕様が適切で,また,装置を運転するのに必要なサー
ビス及びインフラストラクチャーを利用できることを確実にするため,装置の仕様をレビューするとよい。
装置仕様を作成するとき,次の事項を考慮するとよい。
a) 定期的に補充される貯蔵タンクからEOを滅菌器に供給する場合,タンクは,分析のためのサンプル
採取手段,EOのタンクを空にする手段,及び汚染又は重合物の過剰な蓄積時にクリーニングする方
法を備えるのがよい。
b) 液状のエチレンオキサイドが滅菌器に導入されないように,滅菌器にEOガスを導入するシステムに
は気化器を備えているのがよい。
c) ガス状のEOが生成されていることを立証するために,気化器から滅菌チャンバへ入るEOガスの温
度を測定するとよい。
35
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
d) 水蒸気は負荷に湿気を与えるために用いられるものであり,滅菌剤を意図したものではない。供給す
る水蒸気の一貫性は,ボイラへの供給水又は水蒸気の凝縮水を定期的に分析して測定するとよい。
e) チャンバの温度を測定するには,最低2個の温度プローブを使用するのがよい。大容量のチャンバは,
チャンバ全体の温度を反映するデータを保存できる監視/制御システムを確実に動作させるため,よ
り多くの温度プローブを取り付けることがある。
注記 二つの温度センサプローブを用いる目的は,一つのセンサによるプロセスの誤った値を受け
入れてしまうことを防ぐためである。二つの温度センサの温度を比較することで,一つのセ
ンサの故障を検出できる。二重素子の温度プローブの使用は,この要求に適合している。
f)
プロセスの間,常に滅菌器チャンバ内を均一の状態に保つことは重要である。これは,強制ガス循環
によって達成できる。ガス循環システムを用いる場合は,循環が無効である場合には,そのことを示
す監視装置を備えるとよい。それには,ファン又はポンプの電源が入っていることを監視する装置だ
けでは,不十分である。
g) EO又はEO混合ガスのボンベ,タンク又はカートリッジの保管に使用するエリアは,施錠し,換気設
備を備えるとよい。
h) 周囲の環境条件が,EOの供給者によって推奨される温度範囲から外れるところでは,EO容器の保管
エリアに温度制御する設備を設けるのがよい。
例えば,湿度センサのような制御及び監視する装置を実際のプロセス条件下で校正することができない
場合がある。これらの装置の校正結果は,適格性の確認検討に関連付けるとよい。プロセス条件は,例え
ば,湿度センサのような幾つかのタイプのセンサに弊害を与える。センサは,一般に感知素子として用い
られる材料の不可逆な劣化のため,プロセス条件に繰り返しばく露した後に交換が必要な場合がある。こ
れらのセンサは,センサ製造業者/供給者の推奨する保守の回数よりも多い頻度で,保守計画を実施する
ことが必要となる場合がある。
D.6.3.3 指針はない。
D.6.3.4 指針はない。
D.6.3.5 制御又は監視機能の検知しない障害がある場合,滅菌負荷はその要求されるプロセスパラメータ
に適合しないのにリリースされる可能性がある。この誤ったリリースを防ぐため,多くの重要なプロセス
パラメータに対して重複してセンサを設置するのが一般的である。これらの重複するセンサを用いての一
般的な選択肢は次の事項がある。
a) 一つのセンサを制御,もう一つのセンサを監視及び報告用に用いる。
b) 監視及び制御の両方について二つのセンサ又はそれらの平均値を用いる。このシステムは二つのセン
サ間の差が定義した値を超過した場合には,自動的に許容外の条件となる必要がある。
c) 監視及び制御の両方についての二重素子のセンサを用いる。このシステムは二つの素子間の差が定義
した値を超過した場合には,自動的に許容外の条件となる必要がある。
D.7 製品の決定(Product definition)
D.7.1 一般
D.7.1.1 製品の決定は滅菌する医療機器(すなわち,新規又は変更した製品)についての基本情報の文書
化を含む。
医療機器の製品の決定は,医療機器(それ自体),その医療機器を入れる無菌バリアシステム,及び包装
システムに入れる全ての附属品,使用説明書,又はその他の物品を含む。また,医療機器の意図した性能,
36
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
適用可能な製造及び滅菌プロセスの記載も含む。製品の決定のプロセスは,その製品が新しい設計か,又
は製品ファミリの一部かどうかも考慮するとよい。
製品の決定の一部として,次の事項を考慮するとよい。
a) 医療機器の物理的属性(構成及び形状)
b) その医療機器の意図した用途
c) その医療機器は単回使用又は複数回使用のいずれを意図しているか
d) 滅菌プロセスの選択に影響を与える可能性のある設計特性(例えば,バッテリー,光ファイバー,コ
ンピュータチップ)
e) 微生物学的品質に影響する可能性がある原材料/製造条件(例えば,天然物由来の材料)
f)
要求する無菌性保証水準(SAL)
g) 包装
h) 載荷形態:特定の負荷又は混合載荷形態,又は許容できる載荷形態の範囲
i)
EO又はガス混合及びプロセス条件(プレコンディショニング,滅菌及びエアレーションプロセス)
との適合性。
D.7.1.2 技術的レビューでは,既存のEOプロセスのバリデーションに用いたバリデートした製品及び/
又はPCDと新規又は変更した製品とを比較するとよい。新規又は変更した製品の構造及び形状について,
EO,熱又は湿気の浸透の妨げとなる可能性のある全ての特性を注意深く調べるとよい。医療機器製造業者
にとってこの比較には,製造施設の立地,用いた原材料のタイプ,それらの材料の素材及び製造法を含め
て,製品上の初期バイオバーデンに影響する可能性のある要因の調査も含めるとよい。変更した再使用可
能な製品に対しては,この比較には,製品の洗浄効果の評価を含めるとよい。
滅菌特性が既に知られている既存の医療機器又はPCDと新規又は変更された製品とが同等であると立
証される場合,新規又は変更した製品は製品ファミリ又は処理カテゴリの一部分であると考えられる場合
がある。
注記 以前にバリデートした製品/PCDより滅菌サイクルに対して滅菌しにくい新規又は変更した
製品を導入するリスクを最小とするために,AAMI TIR28 [26]は有用な指針である。
滅菌対象製品の形状,密度,又は載荷形態及びその包装が,以前にバリデートした製品より滅菌しにく
い可能性のある場合は,EO,熱及び湿気の浸透の検討及び/又はサイクルの致死率の検討を行うのがよい。
技術的レビューの一部として次の質問を考慮するとよい。次の質問事項のいずれかが“Yes”の場合,以
前にバリデートした製品より滅菌しにくいかどうか判断するために,新規又は変更された製品の詳細な評
価が必要な場合がある。
a) 以前にバリデートした製品と照らし合わせて,新規又は変更した製品は
1) 通路又は内くう(腔)はより狭く閉ざされているか
2) 開放部分は少ないか
3) 内部表面はより大きいか
4) 重なった表面エリア及び/又は閉塞空間はより大きいか
5) より多くの栓/封止があるか
6) 内くう(腔)はより長い,又は狭いか
7) 熱,湿気,又はEOの移動を少なくする可能性のある変更又は違いがあるか
8) バイオバーデン数又はバイオバーデン抵抗性が参照製品の抵抗性より著しく高いか(製造条件,取
扱い,洗浄プロセス又は用いた材料による。)
37
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9) 提案されたプロセス又は滅菌方法によって悪影響を受ける可能性のある材料又は構造を含むか
b) 以前にバリデートした製品と照らし合わせて,新規又は変更した製品の包装は
1) 取扱説明書,又は保護バリアを含めて,包装要素においていずれかの変更があるか
2) 追加の不浸透性の保護バリアはあるか(例えば,EO又は湿気の浸透又は除去を制限又は妨害する
可能性のあるコンテナ,ケース,テンプレート)
3) 包装材料の空隙率の変更はあるか(例えば,紙の坪量,接着剤又はコーティングの処理)
4) 通気材料又は開口部の表面積の潜在的な減少はあるか(例えば,テープ又は第2ラベルの適用,ラ
ベルのサイズ変更)
5) 製品のバイオバーデンレベルの増加があるか
6) バリア層の数の変更があるか
c) 以前にバリデートした製品と照らし合わせて,新規又は変更した製品の載荷形態は
1) 参照負荷のバリデートした載荷形態と著しく異なるか
2) 吸収性材料の量が著しく異なるか
3) 参照負荷の密度と著しく異なるか
4) 負荷容積が著しく異なるか
D.7.1.3 閉塞した空間又は重なった表面のいずれかがあるものについては,後に実施する致死率測定の適
格性の確認検討に用いる内部PCDを指定する際の評価において考慮するとよい。
D.7.1.4 滅菌医療機器の無菌バリアシステムの主な機能は,使用するまで製品の無菌性が保たれているこ
とを保証することである。滅菌処理の間,無菌バリアシステムはプロセス条件に耐え,製品の品質を保証
するために損傷を受けないものである必要がある。
滅菌する製品用の包装システムを選定する場合,重要な設計及び製造ファクターをその滅菌プロセスに
関して考慮する。EOの浸透を確実にするために,滅菌環境に対する包装の浸透性は最も重要である。空
気の除去はEO滅菌プロセスの一部分であるので,その包装システムはまた,シールの完全性へのダメー
ジ又は破袋なしに,ガス注入及び排気時の圧力変化の際,気体が包装内外に出入りできるのがよい。
通常の取扱い及び流通の間,製品を守る無菌バリアシステム(SBS:sterile barrier system)の能力を立証
するとよい。そのSBSが製品を守る能力を失うことなしに滅菌プロセスに耐えることができる証拠を示す
とよい。SBSのバリデーションは,SBSがEO滅菌プロセスにばく露され,潜在的なストレスにさらされ
る可能性があることを考慮するとよい。考慮には真空/圧力水準,圧力変化の速度,温度などを含めると
よい。複数回の滅菌プロセス(D.7.2.1及びD.7.2.2参照)にSBSをばく露することによってSBSの適合性
を立証することが一般的である。
包装の考慮事項はJIS T 0841-1及びJIS T 0841-2に詳細が示されている。
D.7.1.5 チャンバ内の載荷形態は,製品の温度,湿気,EOの浸透及びEOの除去に影響を与える。プロ
セス中の適切な製品温度,湿度及びEOの浸透及びEOの除去を確実にするためバリデーション中に,載
荷形態を定義する。
D.7.1.6 PCDは微生物学的チャレンジを入れた機器である。同等性の立証に用いるためのPCDを開発す
る方法の例は次のものがあるが,これらに限定しない。
a) シリンジストッパーのリング,ランド,グロメット又はリブの間に微生物学的チャレンジを配置した
もの
b) 製品の完全性を復元するために接着剤又はコネクタを用いて再接続されるチューブの内くう(腔)の
中央に微生物学的チャレンジを配置したもの
38
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
c) かん(嵌)合部に微生物学的チャレンジを配置したもの
d) 組み合わせた封筒又は包装に微生物学的チャレンジを配置したもの
幾つかのPCDの設計品は,ヘルスケア施設で用いるために推奨されている。
注記 詳細な情報はANSI/AAMI ST41 [22]参照。内部及び外部PCDの詳細はD.8.6も参照。
内部PCDの作製のために,微生物学的チャレンジは直接又は間接的のいずれでも製品上に接種できる。
直接接種は製品上に芽胞の懸濁液を接種する。間接的接種は,包装内又は製品の中/上のいずれかに接種
担体を置く。
次の事項はPCDを作製する種々の方法である。
a) 接種製品:滅菌する製品に直接的又は間接的に接種してPCDを作製する。
b) 接種した模擬製品:模擬製品は,直接又は間接に接種してPCDとして作製する。模擬製品は,製品フ
ァミリ内の全ての製品を適切に代表し,そのプロセスに対して最も滅菌しにくいものとして知られて
いる医療機器の一部又は部品の組み合わせから構成される。
c) 接種した担体:包装,試験片又はチューブに直接又は間接に接種してPCDを作製する。
注記 芽胞懸濁液の直接接種は,表面現象,他の環境因子,及び製品の上又は中の芽胞による閉塞の
ために,接種された製品の抵抗性の変化をきたすことがある。それゆえ,直接接種の実施は,
接種した製品の抵抗性が日常生産する製品と合理的に関係付けがされていることを確実にする
ように,科学的根拠又はバリデーションを示すことが重要である。抵抗性を平板計数法によっ
て測定する場合は,接種した細菌の回収率もバリデートするとよい。追加情報としてGillis及
びSchmidt [30],West [40]及びJIS T 11737-1を参照。
以前に適格性を確認した製品又は内部PCDとの同等性を立証する方法は,新規又は変更した製品と以前
に適格性を確認した製品/マスタ製品(D.8.6及びD.12.5.2参照)の両方を部分サイクルにばく露したとき
の滅菌しにくい位置に置かれたBIの不活化速度を相対的に比較することである。同等性評価検討は,新規
又は変更した製品とそのプロセスをバリデートするために用いた内部PCDとを比較するとよい。この比較
にPCDを用いる場合,このPCDの抵抗性を年1回レビューの一部として評価するとよい。
D.7.2 製品の安全性,品質及び性能
D.7.2.1 予期している滅菌条件の範囲でEO及び/又は全ての希釈剤による化学的及び物理的変化を許容
できる材料を選択することは重要である。製品の性能に対する要求事項を満足させるのに必要とされる,
物理的強度,通気性,物理的な寸法及び弾性などの材料の特性は,その材料が使用可能であることを確実
にするため,滅菌後に評価する。滅菌プロセスにばく露することによるひび割れ及びぜい(脆)化のよう
な劣化影響を考慮することが必要な場合がある。該当する場合,複数回の滅菌プロセスへのばく露に対す
る影響を評価することも必要な場合がある。
あらかじめ定めた滅菌プロセスがその製品の正しい機能に影響しないことの立証は,医療機器及びその
包装システムについての機能試験又は他の適切な試験を実行することによって成し遂げられる。これらの
試験は,滅菌器内又は他のあらかじめ定めたプロセスを模擬した環境チャンバ内でのばく露後に行うこと
ができ,単純な目視検査から一連の専門的な試験に及ぶことがある。
次は,安全性,品質又は性能に影響する可能性のある項目である。
a) 無菌バリアシステムのシールの完全性に影響を与える可能性があるサイクルの圧力変化
b) EOばく露時間,温度,湿度の影響,及び意図した滅菌器内に混合する全ての希釈ガスの影響(該当
する場合)
c) EO残留物を多く保持することが知られている新しい材料の含有
39
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
d) 包装の特性
e) 潤滑剤の存在,特に重なった表面の内部
f)
分解又は洗浄が要求される医療機器かどうか
g) 安全性に関するハザード(例えば,溶出性材料,バッテリー又は漏れ若しくは爆発する可能性のある
密封された液体)
h) 滅菌サイクルの回数
発火の潜在的な原因(例えば,バッテリー)を含む医療機器は,そのサイクルの全ての部分で爆発範囲
内のEO混合ガス域が含まれないプロセスを用い滅菌するとよい。
D.7.2.2 製品を複数回の滅菌サイクルにかけることの評価は,その製品/包装に対する日常滅菌プロセス
を利用して実施できる。製品の材料,機能及び安全性に関する繰返し滅菌及び必要な全ての前処理の影響
を評価するとよい。
再使用可能医療機器では,製造業者の再生処理指示書(取扱説明書)を入手し,それに従うとよい。こ
の指示書には,そのプロセスの推奨される滅菌パラメータ及び再使用可能医療機器をばく露できる滅菌サ
イクルの回数の限度が記載されている。適用可能な場合,滅菌後の再使用可能医療機器の機能を評価する
ために試験及び検査を行うとよい。その医療機器の製造業者が記載する許容できるサイクル回数は最大回
数であると考えるとよい。最大サイクル回数に至った場合,通知されるようなシステムが整っているとよ
い。
注記 更なる情報はISO 17664参照。
D.7.2.3 指針はない。
D.7.2.4 EOプロセスの後,医療機器のEO残留物を管理するために,適切なエアレーションは必須であ
る。EO除去が最もしにくい位置を考慮し,負荷内の残留物製品試験サンプルの配置を考えるとよい。
製品の残留物がJIS T 0993-7の要求事項に適合している場合でも,局所環境,安全及び衛生に関する規
制は,EO滅菌された製品の取り扱いの際,作業者への特別なばく露予防策を要求する場合がある。
ヘルスケア施設:医療機器のエアレーションに関する情報が,その製造業者から入手できない場合,ヘ
ルスケア施設は,製品及びその材料並びに設計についてのデータ又は知識を用いてその機器のエアレーシ
ョンプロセスを確立するとよい。そのエアレーションプロセスは,最もエアレーションしにくい製品又は
製品ファミリを基に設定するとよい。
D.7.3 微生物学的品質
D.7.3.1 エンドトキシン試験の指針は,ANSI/AAMI ST72及び該当する薬局方に示される。
D.7.3.2 ヘルスケア施設では,微生物学的品質への注意は,使用された再使用可能医療機器の回収及び取
扱いのための厳密な手順,並びに再使用可能医療機器の医療機器の製造業者の再生処理指示書に従った洗
浄プロセスのバリデーション及び管理のための厳密な手順で構成される。
バイオバーデン法(附属書A参照)を用いる場合,バイオバーデン試験は少なくとも四半期ごとにする
とよい。監視の間隔は,次を考慮した文書化したリスク分析に従って,間隔を広げることができる。
− 製品ファミリの使用
− 過去のデータ
− 統計的分析
− 製造頻度及び製品設計
D.7.4 文書化
製品の決定の完了時に次の事項を文書化するとよい。
40
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) 製品形態及びEOプロセスへどのように供するか(包装及び載荷形態)の記載。その仕様は,また,
プロセスへの製品の適合性の証拠又は評価と同様に,要求するSALを記載するか又は参照するとよい。
b) 新規又は変更した製品と既にバリデートした製品との比較の結果。この結果は,製品の複雑さ,材料,
包装及び載荷形態を評価したことを明確に立証するとよい。
c) その製品のバイオバーデン及び内部PCDと比較したその抵抗性についての証拠又は評価。
d) 新規又は変更した製品を,あらかじめ定めたSALを達成するための現在のバリデーション検討におい
て特に参照される製品ファミリ/処理カテゴリに受け入れることが適切であるという,文書化された
結論。この結論には,(既存のバリデーション検討を補うための追加の試験の全ての結果及び既存のバ
リデートされたサイクルでの)製品の日常的なリリースに対して確認/適格性の確認のために行う試
験(すなわち,残留物試験及び機能試験)の全ての結果を記載するか又は参照するとよい。
この文書は承認,保存し,参照できるとよい。
D.8 プロセスの決定(Process definition)
D.8.1 指針はない。
D.8.2 プロセスの決定の結果は詳細な滅菌プロセスの仕様である。
医療機器に用いる滅菌プロセスの選定には,次の点を含めてプロセスの効果に影響する全ての要素の検
討をするのがよい。次を考慮に入れるとよい。
− 滅菌器の適用性
− 適用する滅菌器で実施できる条件の範囲
− 既に他の製品で用いている滅菌プロセス
− 用いる滅菌剤(すなわち,100 %のEO又は希釈ガスと混合されたEO)
− 製品限界(すなわち,温度,湿度,圧力に対する感受性)
− EO残留物及び/又はその反応生成物の限度の要求事項
− プロセス開発実験の結果
プロセスの決定中に製造業者は,微生物学的試験及び医療機器の適切な滅菌プロセスの設定の助けとな
る他の分析ツールを用いる。
確立すべき滅菌プロセスパラメータは,次の事項を含む。
a) プレコンディショニング室内の温度範囲(行う場合)
b) プレコンディショニング室内の相対湿度範囲(行う場合)
c) プレコンディショニング室内の時間設定及び範囲(行う場合)
d) 滅菌チャンバ内の真空及び圧力の水準並びに圧力の変化の速度
e) 行う場合,チャンバ内循環が滅菌剤保持時間中運転していることの確認
f)
滅菌チャンバ内の設定温度及び範囲
g) 滅菌チャンバ内の湿度の制御設定値(圧力又は%RH)及び範囲
h) EO及び希釈ガス(行う場合)の設定導入圧力及び範囲,滅菌チャンバにEO分析装置が据え付けられ
ている場合は,通常EO濃度も含める。
i)
EO保持時間
j)
滅菌チャンバから負荷の取り出しの前のチャンバ内ガスのフラッシングのための設定(行う場合)
k) エアレーション室内の設定温度及び範囲(行う場合)
l)
エアレーション室内の設定時間及び範囲(行う場合)
41
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
m) 空気の流れ/換気回数
注記 附属書A及び附属書Bは,滅菌プロセスの開発における,サイクル致死率の測定のための要求
事項を示す。
ヘルスケア施設のためにヘルスケア施設で再生処理される再使用可能医療機器について,製造業者はバ
リデートしたプロセスの決定の一部分を基礎とした再生処理に関する指示書の提供を求められている。こ
の文書をレビューし,自らの装置及び滅菌プロセスを用いて医療機器の製造業者の指示書に従って処理が
できることを確認するのは,ヘルスケア施設の責任である。ヘルスケア施設の購買手順は,EO滅菌でき
る医療機器の購買の前に,その医療機器が,その施設で用いている滅菌器及び滅菌プロセスに適合してい
ることを確認するために,再生処理指示書を評価するとよい。ISO 17664も参照。
医療機器又は包装材料の製造業者が供給する再生処理指示書が十分ではないか,又は適切ではない場合
(例えば,そのヘルスケア施設がEO及び希釈ガスの混合を用いている場合での100 % EOでのEOプロセ
ス),その施設は,材料への影響データ及び他の医療機器の再生処理指示書を基礎として,バリデーション
するか,又は自らの再生処理方法の適切性を評価するとよい。ヘルスケア施設は製品をバリデートできな
い場合,又は自らの再生処理方法の適切性を評価できない場合,その医療機器を再生処理しない方がよい。
D.8.3 研究用チャンバは,一般的に製造用チャンバより小さい容器であり,バリデーションを補助する実
験に用いることができる。
研究用チャンバの使用は,製造用チャンバでのPQの確認を妨げない。
D.8.4 プロセスの決定を確立する場合,選定したプロセスパラメータ,その許容範囲の製品及びその包装
の安全性,及び機能に対する影響を考慮することは重要である。
滅菌プロセスのパラメータの数は多くあるので(温度,湿度,圧力変化/速度,EO濃度及び時間),全
ての変数の全ての組み合わせの許容範囲を評価することは実際的ではない。どの変数が最も重大な影響を
与えるか決定し,これらを評価するとよい。
この活動を裏付けるデータは代替実験から収集できる。例えば,製品及びその包装バリデーション,製
品及びその包装の安定性試験実験,加速劣化実験など。代替として,研究又は製造用チャンバでの特定の
チャレンジとなるサイクルから収集できる。
D.8.5 指針はない。
D.8.6 BIが適切であることを示すために,幾つかの方法を用いることができる。
アプローチ1
この方法は製品上に認められるほとんどの微生物は参照微生物より低いチャレンジを示すということを
理由として用いられる。この方法は次の場合に適切である。
a) PCD内に用いるBIはISO 11138-2:2006の箇条5及び9.5に従った場合。
b) 製品のバイオバーデンは一定であり,高い抵抗性の微生物を含んでいない場合。
この方法では,バイオバーデン傾向データを利用でき,微生物の数及びタイプについてのバイオバーデ
ンの一貫性を立証するとよい。バイオバーデンの潜在的な発生源を識別し及び管理していることを確実に
するために,製造プロセス及び製品に接触する材料を評価するとよい。
アプローチ2
この方法は,部分サイクルの後に行う製品及びPCDの無菌性の試験の利用である。この実験の結果は,
製品及びPCDの無菌性の試験の生残データを用いた致死率の比較の手段を提供するとよい。
通常この方法において,無菌性の試験で全ての製品は陰性となり,BI/PCDでは生残が認められることを
意図した部分サイクルに製品及びBI/PCDをばく露する。
42
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
アプローチ3
この方法は,次の場合に適用できる。
a) 製品バイオバーデンのチャレンジが,PCDの中のBIのチャレンジと同等以上
b) 製品バイオバーデンが,高い抵抗性をもつ微生物を含む,又は
c) PCD内で使用するBIの菌数が,ISO 11138-2:2006の9.3で要求されるより低い。
この第3の方法では,バイオバーデン及びPCDの致死率のチャレンジ(比較)は,直接計数法及び/又
はフラクションネガティブ法に基づき可能である(ISO 14161:2009参照)。
製品のバイオバーデンのチャレンジが,BIのそれを超えることを示す場合(すなわち,BIが不適切な場
合),次のうちの一つを用いることができる。
a) PCD内で使用するBIは,菌数の多い及び/又は抵抗性の高いものを選択する。
b) バイオバーデン数を減少させるために滅菌前に製品を前処理する。
c) バイオバーデン数又は抵抗性を減らすために,製品,プロセス又はその両者を評価する(例えば,使
用する原材料又は製造プロセスの変更,製造環境の改善,又は製品の設計の変更)。
d) 新しいPCDを開発する。
上記のいずれかに変更した場合,変更の有効性を検証することが大切である。
製品の設計によっては,BIを最も滅菌が困難な場所に置くことができない場合がある。この場合,最も
滅菌が困難な場所との関係が確立できる場所にBIを置くのが適切な場合がある。さらに,多くの医療機器
において,最も滅菌が困難な場所は微生物の数が少なく,したがって,チャレンジ菌数は製品のバイオバ
ーデンにより近い関係かもしれない。
異なったタイプのPCDがD.7.1.6に記載されている。BIの適切性を決定するのに使用されると同様の方
法を,PCDの適切性を測定するのに用いることができる。出荷包装又は出荷包装ケース内の製品内に置か
れるPCDは内部PCDと呼ばれ,出荷ケース及び滅菌負荷の外部表面上に置かれるPCDは外部PCDと呼
ばれる。内部PCDは日常の製品リリースに用いることができる。しかしながら,外部PCDは,滅菌プロ
セス完了後に簡単に取り出すことができるので広く使用されている。研究用のチャンバで実施した検討は,
検討中の内部及び外部PCDの相対的な致死率チャレンジを立証するのに使用することができる。しかし,
これらの実験を実施するとき負荷の容積及び製造用滅菌器の性能の影響を考慮するのがよい。研究用のチ
ャンバが,生産プロセスを再現できない場合,致死率チャレンジの比較実験を製造用滅菌器で実施すると
よい。
内部と外部PCDとの致死率のチャレンジの比較は,同時に部分サイクルにばく露して評価することでで
きる。結果のデータは次のために利用できる。
a) 滅菌プロセスをバリデートするのにどの内部PCDが適切かを判断するため
b) 候補の外部PCD(すなわち,プロセスの日常監視用)の設計を評価するため
c) バリデートした滅菌プロセスへの受入れについての新規又は変更した製品の同等性の評価をするため
d) 新規若しくは変更した製品又は内部PCDが製品ファミリ又は処理グループのマスタ製品になるかの
判断をするため
次のような場合,PCDの致死率チャレンジを製品の致死率チャレンジと比較せずに,PCD同士のチャレ
ンジの比較を行うことがよい場合がある。内部PCDの適切性が証明されており,外部PCDを日常生産サ
イクル監視のために従来からのリリースに導入しようとするとき,又は他の外部PCDに変更することが望
ましいときによく用いられる。この場合,PCDの適切性を評価する方法は,外部PCDは内部PCDと比較
したとき,同等以上の致死率チャレンジを示すことを立証することである。一般的に,内部及び外部PCD
43
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
の部分陰性結果を比較する場合は1回の部分サイクルの実施によって行われる。外部PCDの致死率チャレ
ンジが内部PCDの抵抗性より小さい場合でも,それが20 %以下ならば,これはPCD内で用いるBIの信
頼度からこれらのPCDは同等と考えられる(米国薬局方EO滅菌用BI)。
注記 滅菌がより困難な形態での内部PCDより,滅菌がより容易な形態での外部PCDが高い致死率
チャレンジを示すことが見いだされることはまれではない。EOが内部PCDより外部PCDの方
がより迅速に除去されるため,その結果,微生物チャレンジに対して短いガスばく露時間とな
ることで理論付けられる。
D.8.7 指針はない。
D.8.8 指針はない。
D.8.9 指針はない。
D.9 バリデーション
D.9.1 一般
D.9.1.1 バリデーションの目的は,あらかじめ定めたプロセスが,要求される無菌性保証水準(SAL)に
適合した製品を恒常的に製造することについて,高い保証の度合いで示すことが要求される証拠を文書化
することである。バリデートしたプロセスで滅菌した製品は,あらかじめ定めた製品の機能及び安全性に
関連する仕様及び品質特性への適合を示すとよい(すなわち,製品適合性の検討を通して)。
滅菌プロセスのバリデーションは,試験開始前に定めた許容基準を含んだ承認された文書(例えば,計
画書)に従って実施するとよい。この文書は滅菌専門家がレビューするとよい。
この箇条で定義するバリデーションの要素は次の事項である。
a) IQ
b) OQ
c) PQ
ヘルスケア施設では,IQ及びOQは認定された職員によって実施される場合もあるが,通常,滅菌器製
造業者によって実施される。微生物学的PQデータは,一般的な負荷について滅菌器製造業者によって入
手できる場合がある。
ヘルスケア施設において,これは次の事項を記載し文書化することを意味する。
a) 実施の必要のあるバリデーションの段階
b) 責任のある個人,部署及び/又は外部の受託者のリスト,及びバリデーションの各段階を実施する方
法
c) バリデーションが成功したとする判定基準
ヘルスケア施設は,このバリデーションの実施について,外部サービスを利用するという選択肢がある。
しかしそれでも,この規格の要求事項に適合するバリデーションを確実にする責任はヘルスケア施設にあ
る。
D.9.1.2 指針はない。
D.9.1.3 指針はない。
D.9.1.4 指針はない。
D.9.2 据付適格性の確認(IQ)
D.9.2.1 装置
D.9.2.1.1 IQをサポートする文書は,装置(附属装置を含め)の物理的及び運転の特性の記載を含めると
44
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
よい。関係文書の例は設計仕様,購買発注書,ユーザ要求仕様書,及び機能設計仕様書を含む。
次の事項は,装置が装置仕様及び要求事項に従って据え付けられたことを確実にするため,適格性を確
認するとよい装置の部品の例である。
a) チャンバ及び扉の構造
b) チャンバ及び配管構造のシール及び接続(すなわち,規定された圧力及び真空を維持する能力)
c) ガスと液体の供給システム(例 空気,窒素,蒸気,EO及び水),用いる場合フィルタを含む
d) 装置及び計器の適切な運転のために必要な,十分で一定な電力を供給する電気供給
e) ガス循環システム(実施の場合)
f)
ガス導入システム
g) ポンプ,ポンプ冷却システム及び配管を含めた真空システム
h) 排気,排出制御,及び排気処理システム
i)
プロセス自動化,安全システム等の,プロセス状況に影響することもあるその他の重要なシステム
j)
温度,湿度,圧力,EO濃度のようなパラメータを監視,制御,表示又は記録する計器の校正(例え
ば,センサ,記録計,ゲージ,及び試験装置)
k) IQのための文書化された手順は,この適格性の確認の各要素をどのように計画し,実施し,レビュー
するかをあらかじめ定めるとよい。
D.9.2.1.2 IEC 61010-2-040に指針がある。
D.9.2.1.3 指針はない。
D.9.2.2 据付適格性の確認(IQ)
D.9.2.2.1 据え付ける装置の立地については,関連する国及び地方の法規を遵守するとよい。
D.9.2.2.2 職員がEOにばく露される可能性について,労働安全衛生のための国及び地方の要求事項をど
のように適用するかについて確認するとよい。
職員の労働安全衛生の保護のため,滅菌器近傍及びばく露の可能性のあるその他の場所のEO又はその
ガス混合物の雰囲気レベルを検知する装置を設置するとよい。
EOの安全性は,次の事項を含む要因の組合せで達成され維持される。
a) システム及び装置の適切な設計,据付及び保守
b) 労働安全衛生及び環境保護についての適用される法規への適合
c) 安全作業の実施をサポートする方針,手順の開発及び実施
d) EOのばく露が起こる可能性のある区域での大気モニタリング
e) 適用できる場合,個人用モニターの使用
f)
職員の訓練
g) 設計仕様,施設の方針及び手順への継続的な適合を確実にするために,装置,職員及びプロセスの定
期的な査察
ヘルスケア施設では,IQは一般的に滅菌器製造業者の責任であり,一方,産業施設では,工場の代表者
と一緒に現場職員によってしばしば実施される。IQが製造業者によって実施されるか,又は第三者によっ
て実施される場合,文書及び購買,装置の据付けの記録の保管及び管理の責任はその施設にある。
D.9.2.2.3 EOの貯蔵条件は,EO製造業者の推奨事項及び全ての適用法規に従うとよい。
D.9.2.2.4 指針はない。
D.9.2.2.5 図面,プロセス・機器ダイアグラム(P&ID)及び概要図解は,実際に据え付けられたものに対
しチェックし,必要ならば更新するとよい。
45
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
装置の図面及び部品リストは,次の事項を含むとよい。
a) 配管及び装置の図解(すなわち,プロセス及び機器のダイアグラム)
b) その他の関係する機械的,電気的図面及びその位置のリスト
c) 重要な装置及び機器のリスト,特に物理的な特性及び製造業者の性能表示がプロセス管理に影響を及
ぼすもの(例えば,正確さ,再現性,寸法及び型式)はファイルに保管するとよい。
d) 制御システムレイアウト,制御ロジックダイアグラム,プログラムリスト及びフローチャート,適用
される場合はラダーロジックダイアグラム及び戦略ダイアグラム(連関フローチャート)のような適
用ソフトウェア(コンピュータ化された測定と管理システム)を含んだバリデーションをサポートす
るプロセス制御ロジック又はソフトウェア文書
D.9.2.2.6 指針はない。
D.9.3 運転適格性の確認(OQ)
D.9.3.1 次の情報は監視,管理,表示,又は記録のために用いる全ての計器について文書化するとよい。
a) 装置の識別
b) 校正計画
c) 各校正の実際に終了した日付,実施者
d) 次の計画校正日
D.9.3.2 EO装置のOQは,プロセス仕様に含まれる運転パラメータ及び運転限度の範囲で装置を運転で
きる能力を立証するために,無負荷又は適切な試験材料を用いて実施する。このパラメータの範囲及び運
転限度は,プロセスの決定(箇条8参照)で定義した滅菌プロセスを含めるとよい。
OQは附属システムに関係する性能も明らかにするとよい。例えば,最低EO導入温度を達成するため
のEO気化器の能力。
システムソフトウェア(例えば,コンピュータ化された測定及び制御システム)は,OQの間に全ての
許容外の状況下で試験するとよい。ユーザはソフトウェアがバリデートされていることを確実にすること
に責任がある。
OQには事前に定義したサイクルを用いるとき次の事項を含めることができる。
a) プレコンディショニングフェーズ
1) 滅菌負荷に占有されるエリア全体にわたった循環空気のパターンを測定するとよい。これは空気換
気回数及び風速測定の計算と組み合わせたスモークテストによって実施できる。
2) 温度及び湿度は,望まれる範囲内の値に保たれていることを立証するのに十分な期間にわたってプ
レコンディショニングエリア全体を監視するとよい。プレコンディショニングエリア全体にわたっ
た多くの位置に分散した温度及び湿度を測定するとよい。
注記 温度及び湿度センサ数の推奨として,表C.1及び表C.2を参照。
b) 滅菌フェーズ
1) EOの代わりに不活性ガスを用いる場合,結果を評価するとき比熱容量の違いを考慮するとよい。
2) 温度/湿度分布:温度/湿度センサは最も大きな温度差を示すような,例えば,加熱されないチャ
ンバ又は扉の部分,及び蒸気又はガス入口の近くのような位置に置くとよい。残りの温度センサは
有効チャンバ容積全体に均一に分散して置くとよい。
注記 推奨されるセンサ数については表C.1参照。
3) 空のチャンバでのOQ試験で記録した温度範囲は,EO又は不活性ガスばく露中の有効チャンバ容積
内で,平衡期間後に,各測定時刻で記録したチャンバ温度の平均の±3 ℃であるとよい。チャンバ
46
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
に負荷を入れてOQ試験を実施したとき,許容誤差±3 ℃は達成できない可能性がある。
4) チャンバリーク速度[真空サイクルについては,減圧下(大気圧以下)で実施するか,加圧サイク
ルについては加圧下(大気圧以上)及び減圧下(大気圧以下)で実施する。]
5) コンディショニングフェーズ中の蒸気の導入による圧力上昇
6) 導入するEOガスの温度は気化器仕様の範囲内又はEOの沸点(大気圧下で10.7 ℃)以上にあると
よい。
7) EO濃度を監視することを意図した,EOの導入による圧力の上昇及び到達速度並びに関連する因子
8) EO除去に使用する減圧の到達真空度及び真空到達速度
9) 空気(又はその他のガス)の導入による圧力上昇及び圧力到達速度
10) これらの最後の2段階の繰返し数及び繰返しでの全ての変動
11) ろ過された空気,不活性ガス,水及び蒸気の供給信頼性
12) 制御の再現性を立証するために複数のサイクルを実施するとよい。
13) ジャケット加熱システムによる適切な温度均一性を検証するために,チャンバ壁の温度検討を実施
するとよい。その検討では,そのシステムを継続して効果的に操作するための定期的な検証比較を
するために,温度プロファイルの測定をするとよい。
c) エアレーションフェーズ
1) エアレーションを実施するとき,プレコンディショニングエリアで推奨したのと同じ方法でエアレ
ーションエリアの温度プロファイルを測定するのがよい。エリアの空気の流速及び流れのパターン
も測定するのがよい。
D.9.4 稼働性能適格性の確認(PQ)
D.9.4.1 一般
滅菌プロセスの有効性及び再現性を立証するために,PQは日常管理以上に厳密な微生物学的試験,及
び物理的試験を含む。通常PQは,IQ,OQ試験が完了し承認されるまでは開始しない。許容基準は滅菌
プロセスパラメータ及び微生物チャレンジへの適合を含めるとよい。PQ活動は文書(例 計画書)に明
確に定義するとよい。PQの要素が,別の組織で実施される場合,それらの組織を関連文書で承認すると
よい。4.1及び4.2参照。
D.9.4.1.1 指針はない。
D.9.4.1.2 AAMI TIR28:2009参照[26]。
D.9.4.1.3 指針はない。
D.9.4.1.4 滅菌時の製品の積載方法をあらかじめ定める際,載荷形態(負荷の構成)及び負荷内の品物の
置き方を考慮するとよい。
通常,定めるべき載荷パラメータは,積み重ね形態,全体の密度,寸法,素材構成及びパレット包装の
使用の有無及びそのタイプを含む場合がある。載荷形態は各々の滅菌器について文書化するのがよい。日
常滅菌がチャンバ満載より少ない製品負荷を含む場合,MPQ及びPPQは最小負荷を組み入れるとよい。
製品の置き場所は,あらかじめ定めるとよい。大きな製造用滅菌器ではパレット又はトート内のケース
の位置を指定することになる。小型滅菌器では,ヘルスケア施設で用いられるので,この製品の置き場所
は,滅菌キャリッジ又はキャリアの上のバスケット,パック及び滅菌コンテナを指定する。
PQ中に用いる製品及び負荷は,少なくとも予想される普通の生産の間の最もチャレンジとなる負荷と
して滅菌が困難なものがよい。その負荷は,日常的に滅菌される製品,又はそれらの負荷に似た特性のあ
る材料を含めることができる。載荷形態の変化は滅菌プロセスの致死率に影響する。許容できる載荷形態
47
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
をあらかじめ定めることは重要である。複数の載荷形態を許容する場合,PQ検討で用いる載荷形態は,
最も滅菌しにくい代表又は最も滅菌が困難なパターンとの関係が分かっているとよい。幾つかの負荷寸法
の変更は重大な影響がないと判断する場合もある。
PQ中,次の二つのタイプの負荷が選択できる。
a) 販売できる製品
b) 販売できない製品又は適切な試験材料
D.9.4.1.5 負荷が外科キット,種々のサイズ及び長さの中空器材,種々の包装,及び多くの異なった材料
(例えば,プラスチック,金属,綿など)を含む種々の質量で構成されるとき,それらの材料は,プレコン
ディショニング及びコンディショニング中に熱するときと同じ挙動をとらないこともあるので,載荷形態
を検証することが重要である。
D.9.4.1.6 最大/最小負荷サイズ(D.9.4.1.4参照)及び製品への影響(D.9.4.1.5参照)の考慮に加え,バ
リデーション用に代表又は最もチャレンジとなる負荷を開発する場合,バリデーション用負荷の構成は,
日常的に滅菌するときの負荷の材料/包材が大幅に変化をする全ての特性を考慮するとよい。
バリデーション用負荷に用いる製品又は代替製品材料は,致死率(すなわち,熱,湿気の浸透,及びEO
ガス拡散:密度)の最もチャレンジとなる一般的な条件の代表となるものがよい。大幅に特性が変化する
吸収性材料,浸透を妨げる硬い材料,密封液体,容器などのような負荷材料が含まれることを考慮すると
よい。
D.9.4.1.7 指針はない。
D.9.4.1.8 PQ中に負荷を再使用する場合,その負荷は次の運転の開始の前にエアレーションにかけ,周囲
環境条件と再じゅん(馴)化するとよい。繰返しの再使用の後,負荷の適切性を考慮するとよい。負荷内
のEO残留物によるBIへの影響がないことを確実にするために,ばく露とばく露との間のエアレーション
を行う。じゅん(馴)化時間が不十分な場合,その負荷は,通常の負荷の条件より暖かい環境にある可能
性がある,又は負荷湿度は通常の環境条件より低いかもしれない。それらの状況のいずれかによって,通
常の製品の代表とならないデータが生成される。高すぎる開始温度は非現実的な高い死滅速度となる。低
すぎる湿度は試験芽胞が乾燥した状態となり,非現実的な低い死滅速度を生じる。また,高すぎる湿度に
よって,環境の露点が製品及び/又は負荷温度より高い場合の結果として生じる環境条件は,その負荷及
び製品の中での結露ができ,それは低い異常な死滅速度を結果としてもたらす。
D.9.4.1.9 指針はない。
D.9.4.1.10 指針はない。
D.9.4.2 稼働性能適格性の確認−微生物学的(MPQ)
D.9.4.2.1 プロセスの決定及び該当する場合IQ及びOQ中に観察した結果は,MPQのためのパラメータ
の設定に用いるとよい。ばく露時間は微生物学的適格性の確認のときに変化させる重要なパラメータであ
る。他のパラメータは,通常の製造プロセスより低い致死率を与えるMPQの保証を示すために調節され
る。例えば,温度,湿度,及び/又はEO濃度を,通常のプロセス範囲より低い設定値に設定して運転で
きる。これによって許容できる致死率を生成することになるあらかじめ定めた範囲内で,観察される全て
の値について保証を与える。
プレコンディショニングエリアに入れる製品をあらかじめ定めた許容される最低値又はそれ以下の温度
の製品を用いて,MPQを実施するとよい。例えば,離れた施設での滅菌のための移送のように,製品の初
期温度が変化する可能性が予想される場合,適格性の確認試験の計画にはこの可能性を反映するとよい。
部分サイクル(部分致死又はハーフサイクル)では,そのサイクルのばく露後のフェーズを短くする,
48
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
又はエアレーションフェーズの前又は短いエアレーションフェーズの後にBIを取り出す必要がある場合
がある。これは,そのサイクルのエアレーション中の負荷内に存在するEOによるBIの“残留殺滅作用”
を最小限にするためである。サイクルのばく露後のフェーズを短くするとき,作業者の安全のような因子
に注意するとよい。ばく露時間を除いたMPQに選択したパラメータはMPQを通して固定のままがよい。
注記 人へのEOばく露についての規制が幾つかの国にあることに注意を払うこと。
D.9.4.2.2 MPQで定義した微生物学的チャレンジは,全ての組み合わせた製品の負荷について,要求する
SALの達成を保証するように設計するとよい。この目的のため,PCD又はEO製品ファミリを代表するワ
ーストケースの製品を用いることが一般的である。
PCDは,滅菌負荷内の製品ケース内に置き,均等に分散させるとよい。しかし,その配置は,最も達成
が難しい滅菌条件となる位置を含めるとよい。用いる位置は温度監視に選ばれた点を含めるとよい。パレ
ットに載せた負荷の位置は,また,評価するチャンバ内の全ての層の可能性を保証するために,パレット
の上部及び下部を含めるとよい。
サンプル数の指針は表C.3参照。
D.9.4.2.3 指針はない。
D.9.4.2.4 プロセスの決定に研究用滅菌器を用いる場合,研究用滅菌器での検討から得られたデータと製
造用滅菌器の検討から得られたデータとの関係の確立を考慮するとよい。チャンバの大きさ及びチャンバ
でのEOの導入と除去に必要な時間のため,微生物学的生残曲線の開発は,製造用滅菌器では常に可能と
は限らない。これらの長いガス導入時間と真空時間とによって,インジケータの微生物の要求する部分的
な生残の回収が制限される。これらの生残曲線は,製造用滅菌器で使用される等価なパラメータ,特にEO
濃度を提供することができる研究用滅菌器で開発できる。研究用滅菌器及び製造用滅菌器で開発されたデ
ータの関係を立証する方法は,物理的プロファイルの比較と負荷密度比較を含む。研究用滅菌器で提供さ
れる滅菌条件を製造用滅菌器での物理的プロファイルと比較するのがよい。研究用滅菌器及び製造用滅菌
器での致死率の比較では二つの滅菌器のEO導入時間と排気時間との違いに気を配るとよい。
研究用滅菌器での滅菌プロセスの開発時に,PCDを最終製品ケースの中,又は日常パターン中に置くこ
とがプロセス開発中のPCDに対してケース内の製品との相互関係を示すために重要である。
D.9.4.2.5 AAMI TIR16:2009の4.3.2参照[25]。
D.9.4.2.6 指針はない。
D.9.4.3 稼働性能適格性の確認−物理的(PPQ)
注記 OQから得られる結果は,物理的PQにおいて評価の必要な特性を明らかにするために用いるこ
とができる。
D.9.4.3.1 これらのPPQの運転の中のいずれかで,無菌性又は製品機能性の要求事項に適合しない場合,
追加の適格性の確認の運転が必要かどうかを決定するために調査を実施するとよい。プロセスパラメータ
が規定した限度内に保持されない場合は,調査を実施するとよい。変更を実施した場合,追加の運転が必
要かもしれない。
D.9.4.3.2 PPQは載荷パターン及び分離したパレットであらかじめ定めた文書化された手順で実施すると
よい。小さな負荷がその区域の全体に重要な影響を与えないような大きなプレコンディショニングエリア
では,種々の載荷状態の実験は実際的でなく,必要性はない。
プレコンディショニングのPPQについての指針は,(滅菌中に行う)コンディショニングの適格性の確
認にも適用できる。表C.1及び表C.2の推奨されるセンサの最低数を参照。
a) 指針はない。
49
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) あらかじめ定めたプレコンディショニング時間(行う場合)に,ばく露後の滅菌負荷の製品温度及び
湿度の範囲を確立し,記録することは重要である。
c) プレコンディショニング(行う場合)から滅菌チャンバへの製品移送の間に,製品温度及び湿度の条
件に影響を及ぼす可能性がある。この影響をPQにおいて考慮することは重要であり,PQにおいて移
送時間は,日常滅菌で用いる最大移送時間を考慮し反映することが通常行われる。
d) 温度及び湿度センサは,無菌バリアシステム内又は滅菌負荷内の単位包装の間に置くとよい。プレコ
ンディショニングを行うとき,その製品はあらかじめ定めた時間の範囲内でプレコンディションする
とよい。プレコンディショニングを行わないとき,その負荷内のその温度及び相対湿度は,そのサイ
クルのコンディショニングフェーズの終わりにあらかじめ定めた限度内であるとよい。
滅菌負荷内の温度及び湿度のプロファイルは,滅菌負荷が,温度及び湿度についてあらかじめ決め
た最低値へ到達するのに必要な時間の間,評価をするとよい。
製品では,例えば,パレットの中央,パレットの端,表面などの最もよく湿度の変化を経験する負
荷の区域内の湿度センサの位置を考慮するとよい。PQでは,湿度センサは,その負荷内の包装内に
置くとよい(可能な場合)。これはそのセンサを無菌バリアシステム内又は単位包装の間に置くことで
実施できる。
e) 指針はない。
f)
パラメトリックリリースを用いる場合,ガス保持フェーズ全体のEO濃度プロファイルを,そのフェ
ーズを通じてどのようにガス濃度が変化するかを測定し評価するとよい。
g) 指針はない。
h) 指針はない。
i)
滅菌負荷内の温度センサは,最大の温度変化となりそうな位置に置くとよい。これらの位置はOQ中
に見いだしたホット又はコールド・スポットに配慮するとよい。負荷内のホット及びコールド・スポ
ットの位置は空のチャンバ内の位置とは大きく異なる場合がある。
PQの間,適切な日常プロセスの負荷温度を保証するために,バリデーションにおいて負荷温度とチ
ャンバ温度との関係に気を付けることは重要である。滅菌チャンバ内でセンサを用い,100 %EO,又
は可燃性混合滅菌剤を用いる場合,温度,及び湿度センサは本質安全,又は防爆設計であるとよい。
これらのセンサは,また,EOといずれの希釈ガスにも機能的に使用可能であるとよい。
j)
エアレーションプロセスの間の滅菌負荷内の温度は,許容される残留量を達成するのに要求される時
間を通して測定するか,又は滅菌負荷の温度が安定するのに必要な時間を通して測定するとよい。
注記 これはMPQ/PPQの完了後の追加実験で設定することができる。
D.9.5 バリデーションのレビュー及び承認
D.9.5.1 指針はない。
D.9.5.2 バリデーションプロセス中に観察された全ての不一致を文書化し,そのバリデーションの結果へ
の影響を調べ文書化するとよい。
D.9.5.3 一般的にバリデーション報告書は,バリデーション計画書に定められた責任者によって承認され
る。
D.9.5.4 バリデーション報告書は,また,次の項目を含むか参照するとよい。
− 滅菌器及び滅菌プロセスの仕様
a) IQ及びOQデータ
b) 全てのPQ運転の物理的及び微生物学的記録
50
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
c) 全ての計器の表示,記録計等は校正され,その仕様内であったことの記載
d) 将来のレビュー及び適格性の再確認の条項
e) バリデーション計画書/手順
f)
使用した文書化した手順
g) プロセス管理限界を含めた文書化した操作手順
h) 失敗が発生した場合,その内容,とった是正処置及び意図したバリデーションへの影響
i)
計画書からの逸脱が発生した場合,この逸脱,バリデーション及びその結果への影響の評価の詳細
D.9.5.5 パラメトリックリリースは,基本的な物理的プロセスパラメータが,定義した負荷内の特定の製
品についてのバリデーション中に確立した仕様に適合した場合,製品が滅菌されたとして製品をリリース
する方法である。パラメトリックリリースは,BI又はPCDの試験は行わず,プロセス記録の文書化した
レビューに基づいている。
相対湿度,EO濃度の両方の値及び許容範囲は,あらかじめ決めた数の日常サイクルのレビューの後に
決定する必要があるかもしれない。この評価期間中,プロセス処理する負荷の日常監視及び管理の一部と
してBIを用いる場合がある。選択した運転回数の正当性を理由付けし記録するとよい。このことはその負
荷の均一性,存在するデータ,季節的な変動又は滅菌の頻度に影響される。
ヘルスケア施設で用いられるEO滅菌器は,製品のパラメトリックリリースが許容される適切な装置で
はないことがある。
D.9.5.6 指針はない。
D.10 日常監視及び管理
D.10.1 指針はない。
D.10.2 10.2[a)〜o)]の指針
a) プレコンディショニングエリアに入れる製品の温度は,あらかじめ定めた最低温度以上,又はあらか
じめ定めた貯蔵状態に合致するとよい。例えば,移送中のような,製品を極端な温度にばく露する場
合,内部温度及び湿度が許容範囲内になるようにプレコンディショニングの前に製品を貯蔵するか,
又はプレコンディショニング時間を延長する場合がある。
注記 製品のプレコンディショニングに入れる,又は貯蔵条件の最低温度はPQ中にあらかじめ定
めるとよい。
b) プレコンディショニング中の温度,及び相対湿度を日常監視する参照位置は,要求条件を達成するの
が最も難しい位置と関連付けるとよい。プレコンディショニングエリアの操作のための監視データは,
製品のリリースのための他のデータと合わせてレビューするとよい。
c) 指針はない。
d) 指針はない。
e) 湿度は一般的に圧力変化の測定によって計算する(AAMI TIR15 [24]も参照)。チャンバの湿度は,通
常チャンバに導入した水蒸気の分圧の測定によって計算する。その際相対湿度値は,蒸気圧表を用い
て,実際のサイクルプロセス温度での飽和蒸気圧とこの水蒸気の分圧との比によって求める。これは
チャンバの空間部の相対湿度を示し,更に負荷,又は他の反応が空間部の実際の水蒸気含量に影響す
るまで正しいことになる。プレコンディショニングからチャンバにもち込まれる負荷のもつ湿気の量
を考慮するとよい。
f)
指針はない。
51
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
g) 強制ガス循環システムは,混合ガスを用いるときに均一状態の維持を確実にするため,及び微生物学
的致死率へ影響する可能性があるガスの層分離を避けるため,特に重要である(D.6.3.2参照)。
h) 指針はない。
i)
EO導入による圧力上昇(ΔP)は滅菌チャンバ内の有効な空間内の間接測定による平均EOガス濃度
を示す。EO濃度は滅菌プロセスの効果に影響する重要な変数なので,EO導入による圧力の上昇を記
録するため別に設けられる第2のシステムは必須と考えられる(詳細はAAMI TIR15 [24]参照)。滅菌
プロセスのEO導入及びEOばく露フェーズの間,EOは製品及び包装材料に吸着し,その吸着は制御
用の測定(圧力差)及び第二測定(すなわち,使用したEOの量の測定又はEO濃度の直接測定)の
関係に影響する。
j)
EO導入時間はサイクル間で変化するので,許容できるEO導入時間の範囲をあらかじめ定めることが
一般的な手法である。
k) 指針はない。
l)
指針はない。
m) EOばく露直後の排気時間はサイクル間で変化するので,許容できる排気時間の範囲をあらかじめ定
めることが一般的な手法である。
n) 指針はない。
o) 指針はない。
D.10.3 BIの生育が観察され,物理的プロセス仕様への不適合を根拠とすることができない場合,分析を
行うとよい。これにはプロセス又は装置の変更,及びPQの繰返しが必要となることがある。
D.10.4 次の指針はヘルスケア施設での適用の方法を示す。
ヘルスケア施設での外部CI:滅菌インジケータテープ,インジケータラベル又は印刷インジケータ用表
示記号は,ヘルスケア施設によって組み立てたそれぞれの包装に貼り付けるか印刷するとよい。外部CI
の目的は処理済み及び未処理の品物の識別である。それらは滅菌のパラメータが達成されたかを立証する
ものではない。インジケータはISO 11140-1に従ったタイプ1であるとよい。
ヘルスケア施設での内部CI:
a) 内部CIは滅菌するそれぞれの包装の内側で使用できる。使用する場合,CIはEO,熱及び湿気の浸透
が最も困難な場所と考えられる包装の部分に置くとよい。これはパックの中心である場合,又は中心
でない場合もある。内部CIは無菌性を検証しないが,手続上の誤り及び装置の機能不全を検出するこ
とを可能にする。EOプロセスの全てのパラメータに反応するCIは有用である。
b) 内部CIは使用時に回収し使用者が観察する。使用者は結果の示す情報の判断のためにインジケータの
性能特性について適切な訓練及び知識があるとよい。
c) インジケータが不適切なEOプロセスを示唆した場合,包装品の中身は使用しない方がよい。その使
用しなかった包装品は,負荷識別及びCIを含めて,その全てを,適切なフォローアップのためにその
処理をした部門に返却するとよい。負荷全体を回収するかどうか結論付けるために,物理的監視,負
荷内全てのCI及び微生物学的監視をレビューするとよい。このレビューの記録は維持するとよい。単
一の未反応,又は決定的ではないインジケータは全負荷が未滅菌である証拠として考慮しなくともよ
い。CIは,不適切な包装,滅菌器への不適切な載荷,滅菌チャンバへの過積荷,滅菌器の故障,滅菌
パラメータの不完全な達成,又はプレコンディショニングの不適切さに伴う問題を示すことができる。
CIの“合格”結果は,インジケータが,設置された場所の品物が無菌であることを証明しない。
d) CIは,ISO 11140-1に従ったタイプ3,4,5又は6であるとよい。
52
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
D.10.5 パラメトリックリリースはBIを使用せず,それに代え物理的プロセスパラメータが全ての仕様に
適合することの立証によって,無菌であるとして滅菌から製品をリリースする方法である。それゆえに,
滅菌プロセスが仕様に合致していることを保証するために,チャンバの相対湿度及びEO濃度の直接分析
のような追加のプロセスパラメータのデータを収集する。
a) 温度測定
温度センサの許容外を見つけ出すことができずに,不適切に処理された負荷を不注意でリリースしない
ことを保証するために,滅菌器内で最低2か所の温度測定が要求される。2か所の温度データ点に違いが
ある場合,許容できる温度差はプロセス仕様内にあらかじめ定めるとよい。制御又は監視用センサが仕様
に合わず調査結果がチャンバの読みが正しいという結論にならない場合,負荷は不合格である。
b) 湿度測定
空間部の相対湿度の直接分析は,水蒸気濃度及び相対湿度値を計算できる電気的センサ,ガスクロマト
グラフィ(GC),赤外線(IR)又は他の分光法が,現時点で,水蒸気濃度及び相対湿度値の計算を示すこ
とが可能な方法である。これらの方法の利点は,コンディショニングフェーズでのリアルタイム表示であ
る。電気的センサはEOガスへのばく露の影響を補正するために定期的な校正が要求され,更に,現在の
ところ感応素子として用いられる材料の不可逆的な劣化のために繰返しばく露した後の交換が必要な場合
がある。
c) EOガス濃度測定
EOばく露中に最低ガス濃度が維持されていたことを立証するために要求される分析頻度は,PQ検討の
間に設定するとよい。EOばく露保持時間を通して監視することは,また,EO濃度がその時間を通して変
化するかを測定するためにバリデーションの一部として行うとよい。この分析の結果は分析した製品と載
荷形態に固有のものである。PQ検討中に実施した分析の結果は,サイクル中はどのような頻度で直接分
析するのがよいかを判断する文書化した仕様となることになる。EO濃度の直接分析を行う場合,最低限,
EOばく露の最初及び最後の部分で実施されるEO濃度の直接分析が推奨される。
コンディショニング中の湿度,及びばく露中のEO濃度の測定及び文書化には,特に注意を払うとよい。
IR,GC,マイクロ波,及び他の同様な技術を用いた直接EO濃度測定をするEOサンプリング機器は,滅
菌チャンバ内のEO濃度の代表となる位置に置くとよい。しかし,この測定は,反応性の影響又は負荷影
響のどのような制約もなしに,全ばく露フェーズを通してチャンバ内のその位置でのEO濃度を示すこと
を理解することは重要である。直接分析の結果の再現性及び精度はPQの間に決定するとよい。日常のサ
イクルでの分析は,許容サイクルのための定められた範囲に収まるのがよい。
EOがチャンバ全体に供給され,負荷の空隙に浸透するために,そのサイクルのEO保持圧フェーズの開
始時にチャンバの濃度を安定させるための平衡化時間を導入する必要がある場合がある。
注記1 電気的センサは1サンプル位置だけのEOガス濃度を測定する。それに対して,計算された
EOガス濃度は,EOガス分子が存在可能な空間(容積)内の平均EOガス濃度を代表する。
EOセンサの動的性能特性,EOガス分子に占有された容積内のEOセンサの位置,滅菌剤が
EO及び希釈ガス分子の両方で構成されているときの層分離の可能性,負荷中でのEOの選択
的吸収及び吸着,及び負荷によって占有される体積などのファクターのために,平均EO濃
度から計算した値は,直接測定した値からかなり異なる可能性がある。
注記2 ヘルスケア施設は日常的にパラメトリックリリースを用いない。
53
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
D.11 滅菌からの製品のリリース
D.11.1 この確認は物理的サイクル変数が滅菌プロセス仕様で規定された許容範囲内であることについ
て,指名された者(又はバリデートした自動プロセス)によるプロセス文書の正式なレビューを含めると
よい。パラメトリックリリースが承認され用いられている場合,あらかじめ定めたプロセスパラメータに
適合することを基に製品をリリースできる。
製品の滅菌に続く日常のリリースでは,紙の代わりに電子的記録の確認に基づく場合がある。同様に,
要求される署名は電子的に行う場合もある。電子的署名,及び記録の使用者はこの種の文書の国家及び/
又は国際的要求事項を認識し,それに合致するとよい。プロセス記録,及びリリースの決定のレビューは,
認定を受けた個人が実施するとよい。
D.11.2 指針はない。
D.11.3 物理的仕様に適合しない又はBI(用いる場合)の微生物の生育を観察した場合,滅菌負荷を隔離
区域へ置き,これらの失敗の原因を調査するとよい。この調査は文書化し,引き続き製品の取扱いは文書
化した手順に従うとよい。
制御又は監視センサが故障した場合,次の事項以外,その運転結果の受入れは不可である。
a) 故障の原因が突き止められる。
b) 残りのセンサのデータが仕様内である。
負荷を再処理すると決定した場合,製品及び包装の再滅菌に対する適合性を立証しておくとよい。繰返
しの滅菌ばく露による製品の機能性とEO残留物及び/又は反応生成物のレベルへの影響を考慮するとよ
い。元の滅菌の記録は再滅菌の記録からトレースできるとよい(7.2.2参照)。
包装システムに対する繰返しばく露の影響が分からない場合,製品は再滅菌前に再包装するとよい。
D.11.4 指針はない。
D.12 プロセス有効性の維持
D.12.1 一般
D.12.1.1 プロセスが要求する製品のSALを達成し続けていることを確実にするために,製品,包装,プ
ロセス及び装置へのあらゆる変更を評価する必要がある。製品及びプロセスについての包括的な変更管理
システムの使用が推奨される。
負荷を滅菌する継続した能力を確実にするため,一般的に監視する一つのパラメータは,その製品のバ
イオバーデンである。バイオバーデンは,JIS T 11737-1に従ってモニターするのがよい。微生物数及び/
又はタイプに大きな変化が認められる場合,負荷を適切に滅菌する滅菌プロセスの能力に与える可能性の
ある影響を評価するとよい。
ヘルスケア施設では,洗浄/消毒プロセスが,引き続き有効であり,これに続いて実施する滅菌プロセ
スの準備として適切なバイオバーデンの減少を与えることを確認するために,この洗浄/消毒プロセスの
有効性に関するデータの定期的な見直しを推奨する。洗浄した機器は最終滅菌の前に清浄度の目視検査を
するとよい。清浄化されてない医療機器は滅菌しないとよい。医療機器を適切に滅菌前に清浄化したこと
を確実にするための方針及び手順を準備するとよい(ISO 17664及びISO 15883規格群を参照)。
ヘルスケア施設にとって,例えば,分解のような,その医療機器に固有であり詳細な再生処理指示書を
医療機器の製造業者から得ることが必須である。機器を清浄化したということを確実にするための方針及
び手順を準備するとよい。
D.12.1.2 要求するSAL及び性能特性をもつ製品を継続して提供するプロセスであることを確実にするた
54
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
めに,滅菌及び監視装置の校正のための文書化されたプログラムが必要である。
D.12.2 装置のメンテナンス
D.12.2.1 効果的であるために,予防メンテナンス活動は,製造業者の推奨と装置の性能に基づき定めた
スケジュールに従うとよい。手順を文書化し,保守要員は訓練するとよい。
日常的に保守すべき装置及び/又は校正すべき装置は次のプレコンディショニング,チャンバ,及びエ
アレーション装置を含む場合があるが,これに限定しない。
a) ガスケット及びシール
b) 監視計器
c) EO監視装置(すなわち,環境及び/又はチャンバ)
d) 扉安全インターロック
e) 圧力安全弁又はラプチャーディスク(破裂板)
f)
フィルタ(定期交換用)
g) 蒸発器/気化器
h) チャンバジャケット再循環システム
i)
チャンバジャケットシステム
j)
聴覚的,及び視覚的警報
k) 温度及び湿度センサ装置
l)
蒸気,及び熱供給用ボイラシステム
m) 排気装置(真空ポンプ)
n) はかり(秤)
o) バルブ
p) 圧力変換器
q) タイマ
r) 記録計
s)
空気/ガス循環システム
D.12.2.2 校正していない,又は正しく保守していない滅菌装置は,滅菌サイクル中のプロセスパラメー
タの誤った記録を生成することがある。このデータを製品のリリースに用いた場合,正しく滅菌していな
い負荷がリリースされることがある。
D.12.2.3 指針はない。
D.12.2.4 保守記録を定期的にレビューし,データによって示された全ての調整をすることが必要である。
D.12.3 適格性の再確認
D.12.3.1 IQのレビューは制御及び監視装置の校正状況が許容範囲にあることの確認を含むとよい。変更
管理及び予防メンテナンスプログラムは,滅菌器への変更を実施してないか,又はそのプロセスに影響す
る可能性のある重大な変更を実施していないことを示す。
D.12.3.2 OQのレビューは,元のOQがその年の間で引き続き有効な結果であることを確実にするために,
装置の性能及びエンジニアリング的な変更の評価を含めるとよい(図D.1参照)。
そのために,装置の定期的な適格性の再確認を実施するのが一般的である。次の事項を含めるとよい。
a) 装置のIQ状況のレビュー
b) 装置性能の傾向の評価
c) プレコンディショニングエリア(用いる場合)の温度及び相対湿度プロファイル
55
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
d) チャンバ温度プロファイル
e) エアレーションエリア(用いる場合)の温度プロファイル
これらの適格性の再確認は,前の適格性の(再)確認以来プレコンディショニング(行う場合),チャン
バ又はエアレーションエリアの性能に重大な変化がないことを示すとよい。これらの試験の結果として装
置の変更が必要な場合,OQの適格性の再確認は繰返しが必要とされる場合もある。
注記 複数の滅菌負荷を入れる大きなプレコンディショニング又はエアレーションルームについて,
装置の重大な変更がない場合,適格性の再確認の範囲は削減することができる。削減した適格
性の再確認の根拠は文書化する。
D.12.3.3 PQのレビューは,その滅菌プロセスが指定した製品に対して引き続き有効であるとの評価を含
むとよい。
考慮する事項は,次を含むが,これに限定しない。
a) 装置のIQ状況のレビュー
b) 装置のOQ状況のレビュー
c) 製品の無菌性に影響する可能性のある製品設計,製造及び包装材料,PCD,供給者,製造エリア又は
施設,載荷形態,又は製造プロセスに重大な変化がないことの確認。
d) 著しい製品バイオバーデンの増加がない,及び/又は滅菌プロセスに対する製品バイオバーデンの抵
抗性の変化がないことの確認。これらの数の増加及び/又は抵抗性の変化は,あらかじめ定めたSAL
に製品を滅菌する滅菌プロセスの能力に悪影響を与える場合がある。
e) 前回の適格性の確認以来,個々の滅菌プロセスを仕様限度内で操作していたことの確認。
f)
製品の無菌性に影響する可能性のある滅菌プロセスに対する変化がないことの確認。
g) プロセス仕様(物理パラメータ)が合致していたのにBI又はPCDの無菌性が失敗したことがある場
合,そのレビュー(適格性の再確認が保証されるかどうかの決定のため)。
このレビューに基づき滅菌専門家は物理的及び微生物学的な適格性の再確認に要求される範囲を決める
とよい。レビュー及び判定は文書化するとよい。
レビューの結果として,適格性の再確認に利用できる三つの選択肢がある。
− フルの適格性の確認 − PPQ及びMPQで構成する。これには,例えば,製品/包装設計又は形
態(新規のワーストケース状態を創り出す),プロセス設計又は装置/サービスに対する重大な変更
がある場合に要求されることがある。
− 物理的又は微生物学的な再適格性の再確認は要求されない。 − 製品,包装,装置/サービス及
びプロセスに変更がなく,許容できる滅菌器性能及びエンジニアリングレビュー,及び日常の滅菌
プロセスがその該当期間で信頼できる状態で運転していたような状況では,次回のレビューまで,
物理的又は微生物学的な適格性確認検討の実施が不要であることを正当とするのに専門的判断を用
いることができる。
− 削減したMPQ及びPPQ − これはある状況において必要となる。例えば,製品バイオバーデン
の抵抗性に対する製品負荷内の内部PCDの抵抗性の適切性が継続していることを検証する,又は定
めた間隔の後,前回の適格性の再確認検討以来,不注意な変更がない証拠を示す。これには通常,
負荷の温度と湿度測定を含む最低1回の部分又はハーフサイクルばく露が含まれる。研究用滅菌器
での部分サイクルも適格性の再確認プログラムをサポートするために使用できる。しかし,製造用
滅菌器の適格性の再確認は製造用滅菌器で実施するとよい。
記録した文書のレビューによって製品又は滅菌プロセスで全ての変化を検証するために,1回のMPQサ
56
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
イクル及び負荷温度,湿度測定(MPQ及びPPQ)を,少なくとも2年に1回実施することが推奨される。
滅菌プロセス仕様を変更した場合,滅菌プロセスの適格性の再確認はJIS T 0993-7に規定したEO残留
物の許容限度値に製品が適合していることの確認も含めるとよい。
上記の全ての場合,その決定と同様にその決定の理由を文書化すること,及び適格性の再確認の次回以
降のレビューの計画を定めることは重要である。
次回の年次レビューへのフィードバック
注記 一つ以上の載荷形態をバリデーションしている場合には,全ての適格性の再認識の活動に反映するのがよい。
図D.1−適格性の再確認ディシジョンツリー
D.12.3.4 適格性の再確認は,軽微な変更の度重なる影響が,滅菌プロセスの有効性を損なっていないこ
とを確認するために実施する。
適格性の再確認には,JIS T 0993-7の製品のEO残留物に適合することの検証を含んでもよい。
不注意なプロセス変化が起きていないことを確認し,元のバリデーションが引き続き有効であることを
立証するために,少なくとも毎年,滅菌プロセスの適格性の再確認の実施の必要性について正式な評価を
することは重要である。
適格性の再確認プログラムは,毎年,元のバリデーションの妥当性を維持するのに必要な性能の変動の
許容できる範囲及び水準を定めるとよい。
D.12.3.5 不適合の根本原因を決定するために,調査を開始するとよい。適格性の再確認の妥当性に対す
る不適合の影響は,評価し,至った判断の理由は文書化するとよい。適格性の再確認に関連する引き続く
処置は,適切な品質システムの監視とともに進めるとよい。
D.12.4 変更の評価
D.12.4.1 次の事項は,適格性の再確認を必要とする場合があるがこれに限定しない。
a) 大規模な滅菌器の修理及び変更(制御装置の交換,大規模な改造又は新規の部品の設置)
b) 構造の変更又は移設
c) 日常滅菌の中での説明不可能な滅菌の失敗
Yes
Yes
No
No
No
Yes
年次レビュー
・装置
・変更
・校正
・メンテナンス
・製品
・設計
・包装
・材料
・製造プロセス
・バイオバーデン
・載荷形態
・プロセス
・逸脱
・プロセス失敗
適格性の再認
識を最近2年
以内に実施
重大な変更
フルの適格性の
再確認
削減した適格性の
再確認
文書化した根拠
変更
57
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
d) 製品の変更
e) 包装の変更
f)
滅菌剤及び/又はその供給方法の変更
g) 滅菌への製品の提供方法又は載荷形態の変更
h) 負荷密度の変更
全ての適格性の再確認で用いた参照負荷について,参照負荷が,変更後の製品/形態の代表であること
を確実するために,加えた変更について考慮することは重要である。
D.12.4.2 製品バイオバーデン数又は抵抗性に影響する可能性のある材料,製造場所又はプロセスの方法
に変更を行った場合,適格性の再確認検討が必要となることがある。その検討は,製品バイオバーデン数
又は抵抗性が,内部PCDの適切性又は要求される製品SALへの達成を損なうような水準には増加してい
ないことを立証するとよい。
D.12.4.3 負荷及び載荷形態の再評価によって,滅菌プロセスの有効性に影響する変更が明らかになった
場合,それらの変更は適格性の再確認検討に組み込むとよい。
D.12.4.4 指針はない。
D.12.4.5 指針はない。
D.12.4.6 指針はない。
D.12.5 同等性の評価
D.12.5.1 プロセスの同等性
プロセスの同等性は,二つ以上の装置によって同一のバリデートした滅菌プロセスを提供できることの
立証に用いる方法である。装置が物理的に同等であることは要求しない。たとえ装置から取得したパラメ
ータが統計的に同等ではなくとも,定義しバリデートしたプロセス限界で運転できるならば,同等である
といってもよい(AAMI TIR28 [26]参照)。
複数の装置間のプロセス同等性は,そのプロセスの適格性の確認をするときに要求する試験の量の最小
化を意図する。滅菌プロセスは一つの装置でバリデートするとよい。残りの装置でIQ及びOQ(9.2及び
9.3参照)を実施している場合,残りの装置では削減したPQが可能である。同等性は,また,幾つかの装
置での削減した適格性の再確認に使用可能である。滅菌プロセスを提供する装置は一般的に,チャンバ又
は部屋及び附属する制御システムで構成される。滅菌処理装置群を一つの製造施設,又は幾つかの施設に
設置しているかもしれない。この装置群は,同一のプロセス条件を提供するために独立して使用が可能で
ある場合,全く同じ設計はもちろん,サイズが異なる又は附属装置に違いがあってもよい。
プロセスの同等性は,微生物学的評価と組み合わせたプロセスデータの分析を通して確立できる。この
プロセスデータは,候補の装置が制御の許容範囲内(例えば,バリデートしたプロセスパラメータを信頼
性よく製品に運用していること)であることを立証するとよい。データの分析によって,プロセスがあら
かじめ定めたバリデートしたパラメータについて許容範囲内で運転していることを確認するとよい。微生
物学的評価は,要求するSALの達成を立証することになる。
D.12.5.2 プロセスの同等性の判定基準
プロセスの同等性は,その装置が同じ施設又は異なる施設に設置されているかどうかにかかわらず確立
することができる。プロセスの同等性のプログラムを設定する前に適合すべき判定基準は次の事項である。
a) 箇条9の要求事項に従った少なくとも一つの既存システムの滅菌プロセスのフルバリデーション
b) 全ての装置を技術仕様要求事項に従って据え付け,それらの要求事項に従って運転したことの立証及
び文書化したIQ及びOQ検討
58
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
c) 全てのプロセスのフェーズの許容範囲を含めたプロセスの決定及び文書化
d) 候補の装置及び既存の装置についてのバリデートした許容範囲に関係するプロセスデータ分析
D.12.5.3 プロセス同等性の決定
一つの装置で運用する滅菌プロセスと他の装置で運用するプロセスとの同等性は,各々の装置のバリデ
ートした同一のプロセスを運転したときに観察したデータの比較によって示すことができる。この比較は,
通常の製造負荷で運転しているときに,その装置が再現性のある必要なプロセスパラメータで運転する能
力があることの評価を含めるとよい。そのプロセスのPQの間に観察したデータもまた用いることができ
る。示されたパラメータ及び許容範囲は,既存の装置での滅菌プロセスのPQにおいて,前にバリデート
したものであるとよい。同等性の評価は,微生物学的評価とともにプロセス分析及び評価の実施を含める。
D.12.5.4 プロセス分析及び評価
プロセスデータの分析は,候補の装置及び既存の装置のバリデートしたプロセスについて実施する。プ
ロセスデータは候補の装置から集めるとよい。これらのデータは,特定の滅菌プロセスのためのパラメー
タの限界値,及び既存の装置のPQで観察した結果と比較するとよい。そのパラメータ限界値は,既存の
装置の滅菌プロセス(この規格で規定する全てのプロセス要求事項を含む。)のための元のバリデーション
で設定している。その仕様,許容判定基準,及びパレット又は載荷形態は元のPQで定めた内容と同じで
あるとよい。同等性の測定で評価した実際のパラメータは,一般的に全てのプロセス仕様の一部分である。
それらの選択したパラメータ及びその選択理由は文書化するとよい。試験データの中央値及び変動の両方
を評価する統計的方法をこの評価に用いることができる。統計的分析方法の例はAAMI TIR15 [24]に記載
がある。その例は説明的例示であり,統計的計算,正規性の要求事項及びデータが正規性試験に合致しな
い場合にとるステップについての指針を提供する。プロセス分析及び評価で設定した許容基準に合致しな
い場合,プロセス同等性は立証できない。
D.12.5.5 プレコンディショニング又はエアレーションエリアの評価
プロセスの同等性の確立の判定基準は,一般的にエアレーションに湿度を適用しないので,これを除外
してプレコンディショニング,又はエアレーションエリアでは同じである。各々の環境内の負荷温度及び
湿度の詳細を比較した評価を実施するとよい。少なくとも,負荷内の温度及び湿度均一性,並びにそのエ
リアの対応設定条件及び変数制御記録とこの均一性の関係を評価するとよい。複数の装置に異なった設定
条件を用いる,又は異なった制御限界値を用いる場合,それらは同等であると宣言できないことがある。
プレコンディショニング又はエアレーションの最終段階で性能データの分析は負荷内がパラメータ限界
(例えば,温度分布,EO残留量など)に適合する結論となった場合,プレコンディショニング又はエアレ
ーションプロセスのプロセス同等性は確立できる。製品EO残留量は候補のエアレーション部屋/チャン
バ/セルで証明するとよい。
D.12.5.6 滅菌チャンバの性能評価
候補装置内の負荷で観察したプロセスパラメータと,既存のプロセスでのPQ又は製造中の運転で観察
したデータが示す内容の比較を評価するとよい。その評価の実施前に,比較する重要なプロセス及び負荷
パラメータを定義するとよい。これらのパラメータは各々の滅菌プロセスで固有であるが,次の事項を含
むことがある。
a) 負荷パラメータ
1) 製品温度−EO保持時間中の負荷内の到達温度及び温度分布
2) 製品湿度−コンディショニングの終わりでの負荷内の到達湿度及び湿度分布
b) プロセスパラメータ
59
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1) そのサイクル中の選択した時点でのチャンバ湿度(例えば,コンディショニングの始め及び/又は
終わり)
このパラメータは直接又は蒸気の導入による圧力の上昇によって測定可能である。
2) そのサイクル中の選択した時点でのチャンバプロセス温度(例えば,コンディショニングの始め,
又はEO保持時間中)
3) そのサイクル中のEO保持時間内に選択した回数でのチャンバEOガス濃度
c) 考えられるその他のプロセスパラメータは次を含む。
1) そのサイクル中の選択した時点での到達真空度及び到達速度(ΔP/時間)
2) 加湿時間及び蒸気導入速度(ΔP/時間)
3) EO導入温度及び導入速度(ΔP/時間)及び使用したEOの量(重さ,濃度,又は圧力)
4) 空気又は窒素の導入速度(ΔP/時間)
プロセスが既存のプロセスパラメータ限界値及び全ての追加の合格基準に適合する能力において,同等
であるかないかを示すために,プロセスデータの分析を用いる。測定したデータは分析及び将来のプロセ
スの同等性の決定のために使用できるようにするとよい。
D.12.5.7 微生物学的評価
微生物学的評価では,部分サイクル又はハーフサイクルは,評価した全ての装置において定めた最低限
の製品SALを達成する能力のある滅菌プロセスを立証するために実施する。
注記 プロセス分析中に用いる運転が部分サイクル又はハーフサイクル及び微生物学的監視を含む場
合,そのデータは,また,この評価に用いることができる。
あらかじめ定めた製品SALを与えることに加えて,追加の要素として,滅菌に供する製品のバイオバー
デンレベルへの影響の可能性がある滅菌場所又は製造場所への全ての変更を含めて評価するとよい。製造
施設と滅菌場所との間の距離が延びると,特にその製品が微生物生育に適した場合は,高いバイオバーデ
ンレベルの結果となることがある。製造環境の差異は,たとえその製品が微生物学的生育に適していなく
ても,既存の適格性の確認をしたバイオバーデンレベルより菌数が多いか又は抵抗性が高い製品の製造に
つながることがある。サイト間で移送する製品で,移送の時間及び季節の影響(温度,湿度など)のよう
な異なった移送条件があるとき,その他の問題を評価するとよい。必要な場合,出荷/輸送条件を模擬的
に定義した条件で製品を保持するとよい。
D.12.5.8 結果評価
評価結果によって複数の装置が同等の性能を発揮できるかどうかを決定する。複数の装置が同等の場合,
既に実施した検討で削減したMPQについての要求事項が満足しているので,詳細な適格性の確認は必要
ない。プロセス分析及び評価,又は微生物学的評価のいずれかでそのプロセスは同等ではないと結論付け
た場合,そのプロセスは“同等ではない”と宣言するとよい,そして,フルのPQを実行するとよい。
D.12.5.9 同等性の維持
同等性の維持は,これらの変更が同等性への全面的な決定を危うくしないことを確実にするために,各
装置,製造プロセス,製品負荷及び滅菌プロセスへの変更のレビューを含めるとよい。このレビューは,
変更前に行うとよく,及び変更管理プロセスの一部分とするとよい。定期的同等性のレビューでいずれか
のプロセスが適合しない場合,それは同等性リストから取り除き,別途に適格性の再確認を行うこととな
る。
D.12.5.10 文書化
候補の装置が既存の滅菌プロセス装置への同等性の宣言ができるかどうかを決めるための分析の結果に
60
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
関連する全ての決定は,文書化するとよい。少なくとも,この文書一式は次の事項を含むとよい。
a) 候補の装置の全てを記載し完成した仕様。それには運転仕様並びに許容範囲,これには適用できる運
転手順,校正手順,及びメンテナンス計画のそれらを参照するか提供するリスト。この仕様は,この
規格に従った前回のIQを含む,又は参照するとよい。
b) 意図したプロセスを与える装置の能力の証拠又は評価。その証拠又は評価は前回のOQを含む又は参
照するとよい。
c) 候補のプロセス装置と既存のバリデートしたプロセス装置との比較の結果。この比較は,全ての重要
なシステム及び重要なパラメータを評価し,統計的分析(用いる場合)を含めていることを明確に示
すとよい。
d) 既存プロセスとの同等性を立証するために,候補の装置内で行うプロセスでの製品の状態の証拠又は
評価。
e) 該当する場合,滅菌プロセスの致死率に影響する可能性のある全ての追加要因の評価の結果。
f)
候補の装置が,あらかじめ定めた製品のSALを達成することを示すバリデーション検討で,明確に引
用された装置と同等性であるとの文書化した結論。この結論は,前回のバリデーション検討を補足す
る全ての追加実施した試験,及び既存のバリデートしたサイクルから製品の日常的なリリースのため
の確認又は適格性の確認の全ての詳細な試験実施(例えば,最初の3ロットの残留物試験,機能試験
など)を含めるか参照するとよい。
g) 滅菌専門家及び他の個人による,通常の変更管理又は組織内のプロセス文書管理手続で要求される承
認
h) 候補の装置を日常の製品処理に用いてもよいと承認するために発行又は変更した適用可能な滅菌操作
手順のリスト及び仕様。
D.12.5.11 製品
D.12.5.11.1 製品ファミリ
製品ファミリは,バリデーションの目的のための類似又は同等であると決定された製品の集まりである。
EO滅菌において,製品ファミリは,他の理由(EO残留物,バイオバーデン又は生体適合性)の目的のた
め用いることもできるが,通常MPQ中に製品へ与えられた要求されるSAL測定の目的のために,一緒に
したグループ製品として参照される。
EO製品ファミリは類似製品の種々の組み合わせを含めることができる。例えば,同じ材質で同じ環境
で作られた製品のサイズ又は品種だけが異なるカテーテルを含んでもよい。製品がファミリにグループ化
されるとき,EO滅菌プロセスへの適切性を根拠としてグループ化することが重要である。
製品ファミリの使用は,それに含まれる全ての製品が,代表製品又は内部PCDよりも滅菌プロセスに同
等又はそれより低いチャレンジを示すことによって,ファミリ内の全ての製品のバリデーションプロセス
をより単純にする。製品ファミリは,ワーストケース製品(よく“マスタ製品”と呼ばれる。)によって代
表することができる。全ファミリは滅菌プロセスに同等なチャレンジである,又はPCD(内部PCD)によ
って代表されると考えられる。
製品ファミリに加えて,処理カテゴリもまたPQが完了するとEO滅菌に日常的に用いられる。処理カ
テゴリは製品ファミリの確立に用いた詳細部を構成する材料及び包装,又は製造業者のような不一致があ
るEO製品ファミリの集合である。各々のEO製品ファミリの処理カテゴリ内の共通滅菌プロセスで適格
性の確認をするとよい。例えば,静脈セットといった製品の集合は製品ファミリを構成するかもしれない。
また,別々の製品の集合(例えば,シリンジのファミリ)を含む処理カテゴリに位置付けられるかもしれ
61
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ない。処理カテゴリ内の共通項目は,グループ内製品の微生物学的代表としてPCDとなることがある。処
理カテゴリ内の全ての製品は,ワーストケース製品,代表メンバ,又は製品無菌バリアシステム内にある
内部PCDと比較したときの滅菌プロセスに対して同等又はより低いチャレンジとなるとよい。
製品同等性のレビューは,製品ファミリ又は処理カテゴリ内で実施することができる。ワーストケース
製品又は代表メンバのいずれか一方は,適格性の確認検討で選定できる。次の箇条は,幾つかの製品評価
の取扱いを示している。
D.12.5.11.2 製品への悪影響の決定
候補の製品又は包装システムが,ある製品ファミリ又は処理カテゴリに取り入れることができるかどう
か決定する前に,候補の製品又は包装システムが機能及び有効性が維持されているかどうか決定する(製
品適格性の確認の実施)とよい。これらを評価するシステムは,設計又は変更管理プロセスで示すとよい。
機能性,完全性,安定性,生体適合性及び残留物を考慮し,更に,医薬品を含む可能性のある機器又は部
品の場合,滅菌プロセスの影響の決定を考慮するとよい。ある種の最終部品を含む製品(例えば,医薬品
入りキット)について,製造業者は,含まれる製品の有効期限に滅菌プロセスが影響する可能性とともに,
構成品についての安全性及び有効性に関する規制要求事項を考慮するとよい。
試験する製品のEOプロセスは,その製品及びその包装システムに対して代表チャレンジとなる構成要
素がよい。このチャレンジが公称プロセスチャレンジとどのように異なるかを文書で示すとよい。また,
製品適格性の確認によって,これらのパラメータにおいて製品が受入れ可であることを立証するとよい。
候補の製品及びその包装は,製品のEO残留量の影響を測定評価するとよい。また,全ての変更の製品
リリースへの影響を評価するとよい。JIS T 0993-7はこの評価の指針とし用いるとよい。
D.12.5.11.3 製品設計の影響の決定
候補の製品の設計は,既存の製品又はPCDよりEO,熱又は湿気の浸透が大きな妨げとなる可能性のあ
る全ての変更又は差異を注意深くレビューするとよい。可能な変更の例は,長い内くう(腔),栓/封止の
追加,又はより多くの接触した表面若しくは製品密度を含む。
製品の機能に対する悪影響とならない変更であることを確実にするために,元の製品機能に対する製品
設計で機能上の試験をレビューする。
注記 この評価は,通常,密封シールされたもの及び意図した使用の間に露出できない機器の部分は
含まない。例えば,シールされたもの,中空部分,成形部品又はシールされた内くう(腔)で
ある。
D.12.5.11.4 製品材料及び特性の影響の決定
候補の製品の特性は,製品バイオバーデンに影響する可能性のある製造方法,施設,場所,原材料のタ
イプ及び素材のような全ての違いについて注意深く試験するとよい。構成材料は,製品が高いEO残留量
又は規制限界を超えない水準を保持していることを確実にするためにレビューするとよい。
D.12.5.11.5 無菌バリアシステムの影響の決定
候補の製品の無菌バリアシステムは,EO,熱又は湿気の浸透の大きな妨げとなる可能性のある全ての要
因を注意深く調査するとよい。その滅菌プロセスに対して候補の製品が既存の製品又は製品内部PCDより,
より大きなチャレンジとなる要因には,通気孔の空隙の減少,通気孔の表面積の縮小,通気孔域の詰まり
又は他の特性を含む。付け加えて,無菌バリアシステムの変更による製品のバイオバーデンへの影響及び
全てのEO残留量への影響を評価するとよい。
D.12.5.11.6 載荷形態の影響の決定
候補の製品の載荷形態は,その滅菌プロセスへの熱力学的な反応に影響する可能性のある全ての変更に
62
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ついて注意深く調査するとよい。候補の製品が滅菌プロセスに対してより大きなチャレンジとなる変更は,
伸縮性のラップの層の追加,パレットの再配置,負荷サイズの変更,負荷の全体の密度の変更,又は他の
全ての変更がある。
D.12.5.11.7 製品ファミリへの適合性の評価の結論
記載された技術的レビューの結果が,候補の製品及び既存の製品又は元の内部PCDは類似している及び
差異は重要ではないと判定される場合,又はバリデートした製品若しくは内部PCDよりチャレンジとなら
ない場合,候補の製品は,製品ファミリ又は処理カテゴリに詳細な検討なしに適合するとしてもよい。
AAMI TIR28:2009 [26]の附属書Aをレビューに用いた場合,質問事項に全て“No”の回答であれば決定が
支持される。この決定の根拠は滅菌専門家によって作成及び文書化されるとよい。技術的レビューによっ
て候補の製品がその滅菌プロセスに現行バリデートした製品若しくは内部PCDより大きいチャレンジと
なる可能性がある場合,詳細な検討が望ましい。候補の製品がその滅菌プロセスに対して大きなチャレン
ジとなる代表として判定された場合,既存の製品ファミリ又は処理カテゴリに合致せず,フルのPQの実
施が必要となる。このPQは次の事項のいずれかで可能である。
a) 候補の製品を代表製品とした新しい製品ファミリ又は処理カテゴリの確立
b) その滅菌プロセスのための新しい内部PCDの確立
c) 候補の製品が現行のバリデートしたマスタ製品に同等であるとの確立
d) 候補の製品の新しい滅菌プロセスの確立
D.13 附属書Aの指針−滅菌プロセスの致死率の決定−バイオロジカルインジケータ/バイオバーデン
法(BI/バイオバーデン法)
D.13.1[A.1] 一般
D.13.1.1[A.1.1] この箇条は附属書A及びD.8,D.9の情報の詳細な指針を提供する。BI/バイオバーデ
ン法及びオーバーキル法は同じ手順で用いられるので,この項の文章の一部はD.14と重複する。
BI/バイオバーデン法は,バイオバーデンよりも同等以上の菌数で抵抗性のあるBI又はその他の内部
PCDの使用に基づく。製品に10−6のSALを提供できるバリデーション検討中に,製品バイオバーデンの
抵抗性及び数が適切に表すことができることを立証するバイオバーデン監視プログラムから十分なバイオ
バーデンデータが得られるとき,この方法は適切である。
注記 この方法は,106以下の菌数のBIの使用又は他の内部PCDの使用を含むことができる。
内部PCDの相対的抵抗性及び菌数は,製品バイオバーデンの抵抗性及び菌数と比較するとよい。内部
PCDの対数減少が,滅菌プロセスに対して最も抵抗性が強いバイオバーデンについて達成される無菌性保
証水準を計算するのに用いることができる。
この場合,BIについての致死率検討での開発で得られた芽胞対数減少(SLR)は,製品についてのプロ
セスの有効性を立証するのに用いることができる。データを計数法によって得る場合,SLRも得られた生
残曲線データから予測できる。ユーザは,この方法から導き出した最低サイクル時間は,それ自身では,
滅菌プロセスをバリデートするためには十分ではないことに注意を払うとよい。提案したフルサイクルの
間,プロセスパラメータが定めた限度で維持できる能力を示すことが必要である。
製品バイオバーデンを頻繁に試験し,それが一定である場合,バイオロジカルインジケータ/バイオバ
ーデンアプローチ組合せ法はプロセスの決定及び/又はMPQに用いることができる。
プロセス致死率の決定:プロセスにばく露後の製品に与える微生物学的致死率は特定の微生物のD値を
基に計算できる。微生物は一般的に与えられたプロセスでおよそ対数的な割合で死滅するので,単位時間
63
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
当たりのEOへのばく露は,菌数にかかわらず微生物の菌数の1/10に減少する。この単位時間は,あらか
じめ定められた滅菌プロセスにばく露したとき,製品の微生物汚染のD値という。
あらかじめ定めた滅菌プロセスにばく露したときの製品に与える特定の微生物のD値及び微生物学的致
死率は,一般的に用いられている二つの方法のうちの一つの方法で結果を計算できる。第1の方法(計数)
は,生残数の計数又は実地計数である。また第2の方法(部分陰性)は部分サイクルの間の生育/非生育
の結果を用いる。これらは附属書A又は附属書Bの方法のいずれかに用いることができる。D値は,部分
サイクルの結果を用いて,ISO 11138-1及びISO 14161に記載された式で計算できる。
EO導入及びばく露後の排気時間の影響を致死率の測定において考慮することは,より高い精度を与え
るので,適切なことがある。EOばく露時間と比較して,EO導入及びばく露後の時間が長いとき,これら
の影響は最も重要である[40]。
用いた方法によらず,次の事項が前提である。
a) 微生物数は均一である。
b) プロセスパラメータはバッチ間で一定である。
c) 片対数生残関係がある。
d) そのプロセスで処理した及び未ばく露微生物は,回収培地で同様な応答で生残している微生物である。
e) 全ての微生物試験法(無菌性の試験,計数など)はJIS T 11737-1及びJIS T 11737-2に従ってバリデ
ートするとよい。
計数法:計数は内部PCDを部分サイクルへのばく露,チャレンジの取り出し,及びサンプル又はBIの
生残数の計数から成る。生残数は生残曲線及びD値に使用できる。D値は直線回帰モデルを用いて計算さ
れる。
ISO 14161:2009参照。
フラクションネガティブ法(部分陰性法):フラクションネガティブ法(部分陰性法)は,滅菌サイク
ルの運転をして全てではないが幾つかのBIが不活化する必要がある。それは次を含む。
a) ホルコム・スパーマン・カーバー(HSK)法
b) 限定ホルコム・スパーマン・カーバー(LHSK)法
c) ストゥンボ・マーフィー・コクラン(SMC)法
ISO 14161:2009参照。
サンプルサイズ:サンプルの数は,用いる方法及びサンプルを負荷に分散しておくか又は1か所に集中
させるかによる。1か所に集中した使用の場合は,サンプル間の結果の整合性を改善することがある。し
かし,広範囲のマッピングを各々のチャンバ,各々可能な載荷形態で実施していない場合は,チャンバ内
のワーストケース位置を代表しないことがある。
結果を評価しているとき,繰返しチャレンジの間,ばく露条件の変動よりも菌数のランダムな変動のた
めに微生物の生残数が異なることがないようにする考慮が必要である。
BIの数の詳細な指針は表C.3参照。付け加えて,サンプルの最低数に適合することを確実にするためISO
11138-1及びISO 14161を参照。
望まれる結果を達成するために,そのサイクルのばく露後のフェーズを短くする必要な場合がある。
D.13.1.2[A.1.2] BIの培養期間の情報は,ISO 14161:2009の12.3に示されている。
D.13.1.3[A.1.3] 致死率又はD値の測定のために計数法及びフラクションネガティブ法を組み合わせる
ことは可能である。二つの方法は異なった計算方法を基にしている。ユーザは一般的にプロセス致死率の
測定のためにいずれか一つの方法を選択する。
64
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
D.13.2[A.2] 手順
無菌性を達成するのが最も困難な製品内での部位は,滅菌剤の浸透が少ない部位だけでなく,多量のバ
イオバーデンが存在するような部位を含む。製品のレビューは,微生物学的チャレンジの適切な設置位置
を確立するために実施するとよい。そのレビューは文書化するとよい。ISO 14161:2009の7.2.2を参照。
考慮すべき点は次の事項である。
a) 内くう(腔)の長さ及び内径,及び医療機器の壁がEOの拡散を許すかどうか
b) 製品及び材料の両方の異なった部品の吸収性
c) 物品の重さ及び密度
d) 載荷形態,特に混載した製品負荷
サンプルの最低数の要求事項に合致することを確実にするためISO 11138-1及びISO 14161を参照。
ISO 14161:2009の附属書Aは,BI/バイオバーデン法におけるBIと製品バイオバーデンとの関係を用
いる場合の追加的な指針を提供する。
内部PCDは,少なくとも製品の最もアクセスしにくい部位のバイオバーデンよりも同等以上のチャレン
ジを提供することが重要である。PCDの開発の情報はD.7.1.6を参照及び製品の無菌バリアシステム内に
置いた内部PCDの適切性の決定の情報はD.8.6参照。
主に致死率に影響するパラメータは,ばく露時間,EO濃度,湿度及び温度である。ばく露時間以外の
パラメータを調整した場合,パラメータには相互関係があり,この調整によって希望した結果を達成でき
ないかもしれないので,そのサイクルへの全体的な影響を評価するとよい。例えば,温度低下の結果は,
圧力パラメータを変更しない場合,実際にEO濃度及び相対湿度の上昇となる。
プロセス致死の検討から得たデータは,滅菌プロセスに要求される最短のEOばく露時間を確立するた
めに使用される。これらの検討が研究用のチャンバで実施される場合,死滅曲線(致死率又はD値/SLR)
はプロセスパラメータ,チャンバ載荷形態,及び検討に用いた負荷内のPCDの場所に固有なので,実際の
滅菌プロセスに対して,このばく露時間を直接適用することに注意するとよい。
直接計数法及びフラクションネガティブ法の追加説明はISO 11138-1:2006の附属書D及びISO
14161:2009の附属書Cを参照。
D.14 附属書Bの指針−安全率を見込んだ滅菌プロセスの致死率の決定−オーバーキル法
D.14.1[B.1] 一般
D.14.1.1[B.1.1] この箇条は附属書Bの詳細な指針情報を提供している。また,箇条8及び箇条9の追
加的指針情報である。BI/バイオバーデン法,及びオーバーキル法は多くの同じ手順を用いるので,幾つ
かのこの附属書の文章はD.13の文章と重複する。
しかし,サイクル計算法を用いる場合,D.13.1.1を参照。オーバーキル法の使用に関する詳細な情報は
ISO 14161:2009の7.2参照。
ユーザは,この方法から導き出された最低サイクル時間は,それ自身では,滅菌プロセスをバリデート
するために十分ではないことに注意を払うとよい。提案されたフルサイクルの間,プロセスパラメータが
定義された限度で維持できる能力の立証が必要である。
D.14.1.2[B.1.2] この方法では一般的に二つの方法が用いられる。
ハーフサイクル法:使用の相対的簡便さ及び安全率を見込んだSALが得られることから,医療機器製造
業者及びヘルスケア施設は一般的にハーフサイクルばく露時間でチャレンジBIの106の全ての不活化を立
証するこの方法を用いる。このばく露時間が2倍のとき,EOばく露中に最低12 SLRが得られる。この方
65
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
法は12 SLR相当以上のプロセスを提供する。
サイクル計算法:この方法は,内部PCDの実験サイクルへのばく露,チャレンジの取り出し及び生残数
の試験から成る。この試験は,フラクションネガティブ手法,又はサンプル上の,若しくはチャレンジイ
ンジケータの生育可能な微生物の計数の実施である。この情報は,製品に定義したSALを与えるために必
要なサイクルの計算に用いることができる。ISO 14161:2009を参照。ストゥンボ・マーフィー・コクラン
法及びオーバーキルサイクル計算法を用いるとき,BI/PCDの推奨される数量は,滅菌される製品容積を
基にして最低10個である。参考文献[38]及びC.3参照。ゼロ時間でばく露するサンプルセットは,実験サ
イクルの滅菌剤導入前の全段階にばく露するとよい。
D.14.1.3[B.1.3] BIの培養期間の情報はISO 14161:2009の12.3に示されている。
D.14.1.4[B.1.4] 製品のバイオバーデン抵抗性に対する相対的なBIの適切性は,適切なばく露時間の部
分サイクルを用いたプロセスの決定の前又は間に実施する無菌性の試験によって立証できる。
D.14.2[B.2] 手順
D.14.2.1[B.2.1] この方法では内部PCDを製品無菌バリアシステム内に置いて使用することができる。
この方法を用いる場合,製品が示すものよりは滅菌プロセスに対して大きなチャレンジを提供することを
示すとよい。内部PCDの滅菌プロセスに対するチャレンジは,少なくとも負荷の中で最も到達しにくい製
品の部位(D.7.1.6及びD.8.6参照)に存在するバイオバーデンと同等であるとよい。PCDの開発について
は7.1.6を参照,製品の微生物学的チャレンジの内部PCDの適切性の判断についての情報は8.6及びD.8.6
を参照。
D.14.2.2[B.2.2] 無菌性を達成するのが最も困難な製品内での部位は,滅菌剤の浸透が少ない部位だけ
でなく,多量のバイオバーデンが存在するような部位を含む。
考慮すべき点は次の事項である。
a) 内くう(腔)の長さ及び内径,及び医療機器の壁がEOの拡散を許すかどうか
b) 製品及び材料の両方の異なった部品の吸収性
c) 物品の重さ及び密度
d) 載荷形態,特に混載した製品負荷
ヘルスケア施設での考慮点:EO,湿度及び熱の製品への十分な浸透を立証するために,EO滅菌プロセ
スの日常監視及びバリデーションのためにPCDを選定するとよい。EOに対するPCDの抵抗性は,製品の
最も滅菌しにくい部位の製品のバイオバーデンの抵抗性と同等以上であることを示すのがよい。
D.14.2.3[B.2.3] 指針はない。
D.14.2.4[B.2.4] 微生物学的計数データ又は部分死滅データの取得には,微生物学的チャレンジを通常
の滅菌サイクルより少ない致死となる条件にばく露することが要求される。これは通常,全てのパラメー
タを一定で通常の状態に保持する,又は許容最低プロセス条件を選定し,ばく露時間を少なくして達成す
る。計数実験で許容最低温度を用いることは,あらかじめ定めた温度範囲内で操作したとき要求される致
死率が得られることを保証する。
致死率に主に影響するパラメータは,ばく露時間,EO濃度,湿度及び温度である。ばく露時間以外の
パラメータを調整した場合,このパラメータが相互に関係があり,この調整によって希望した結果を達成
できないかもしれないので,そのサイクルへの全体的な影響を評価するとよい。例えば,温度低下の結果
は,蒸気導入圧及びEO導入圧力上昇を行わない場合,EOの濃度及び相対湿度は実際には増加する。
D.14.2.5[B.2.5] SLRは部分サイクルの結果を用いて計算できる。内部PCDの生残がない場合,SLRの
ワーストケースの確立は,生残1と仮定した計算を行って得ることができる。
66
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
用いた方法によらず,次の事項が前提である。
a) 微生物の菌数は均一である。
b) プロセスパラメータはバッチ間で一定である(ガスばく露時間を除き)。
c) 片対数生残関係がある。
d) 回収培地内でばく露及び非ばく露の微生物は同様な反応をする。
67
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書E
(規定)
単一ロットの出荷
E.1
一般
この附属書は,単一の滅菌負荷を構成する十分な製品がある場合,例えば,新製品の研究及び開発又は
臨床試験用製品について滅菌プロセスからの製品リリースための要求事項を規定する。
注記 臨床用製品については国の規制のある可能性のあることに注意を払わなければならない。この
ような規制が有効な場合,その規制の要求事項に従うとよい。
E.2
手順
E.2.1 対象の包装された製品が,滅菌する既存の製品ファミリに該当するかどうか評価する。この評価は
製品組成,設計,包装,バイオバーデン及び負荷密度を考慮しなければならない。この評価の結果,決定
に至った理論を含めて文書化しなければならない。
E.2.2 包装された製品が既存製品ファミリに該当した場合,12.5.2及びD.12.5.2参照。
E.2.3 既存の製品ファミリがない場合,又は既存製品ファミリに該当しない包装された製品の場合,次の
事項を実施する。
a) ロットからランダムにサンプルを採り,JIS T 11737-1に従いそのロットの平均バイオバーデンを測定
する。
b) 包装された製品内にある製品無菌性の試験のサンプル及び内部PCDを,滅菌負荷内に滅菌条件を達成
するのが最も難しい位置を含んで配置する。定義した位置に外部PCD(用いる場合)を置く。PCDは
ISO 11138-2:2006の箇条5及び9.5に適合するBIを含む。
注記 位置は温度監視に用いられている位置を含めるとよい。
c) 滅菌負荷を最低プロセスパラメータが推定で製品に10−1未満のSALを与える及び7〜8 log10の減衰を
PCDに与える部分EOガスばく露サイクルにばく露する。
d) 内部PCD,外部PCD(用いる場合)及び負荷からの製品試験サンプルの取り出し及びJIS T 11737-2
に従った無菌性の試験にかける。
注記 内部PCDに対する製品バイオバーデンの抵抗性の比較をE.2.3 c) の部分サイクルより短い時
間の部分サイクルを用いて,以前に評価している及び製品の無菌性の試験サンプルから陽性
試験結果がない場合,E.2.3 c) の部分サイクルにばく露した製品試験サンプルの無菌性の試
験を実施する必要性はない。
e) 負荷を周囲条件へエアレーション及び再じゅん(馴)化する。エアレーション期間は,フルばく露滅
菌サイクル[次のf) 及びg) 参照]内で新しいPCDに悪い影響を与えないことになるレベルにEO残
留物を蒸散させるのに十分である時間とする。
f)
最も達成が難しい滅菌条件の位置を含めて,滅菌負荷内に製品包装内に置かれた新しい内部PCDを配
置する。定義した位置に外部PCD(用いる場合)を置く。
注記 用いられる位置は温度監視に用いられる位置を含めるとよい。
g) 同じ負荷を2回目の公称プロセスパラメータで滅菌サイクルにばく露する。その負荷のあらかじめ定
めたばく露時間は上記c) の部分サイクルの少なくとも2倍である(これはフルサイクルである。)。
68
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
h) 外部PCD(用いる場合)及び内部PCDを再処理負荷から取り出し,無菌性の試験の必要がある。
E.2.4 滅菌負荷は次の要求事項に合致する場合,滅菌から出荷できる。
a) 外部PCD(用いる場合)及び内部PCDに用いられたBIより滅菌プロセスにチャレンジとならない製
品バイオバーデンの存在
b) プロセス仕様に適合する部分サイクル[E.2.3 c)]のプロセスパラメータ
c) 負荷はE.2.3 c) の部分サイクルの少なくとも2倍のあらかじめ定められたばく露時間の公称プロセス
パラメータによってフル滅菌サイクルにばく露再処理されている。
d) フル滅菌サイクルのプロセスパラメータはプロセス仕様に適合する。
e) 部分滅菌サイクルにばく露した外部PCD(用いる場合)及び内部PCDから試験微生物の生育がない
ことの確認
f)
部分滅菌サイクルにばく露した無菌性の試験サンプル製品から生育のないことの確認
注記 内部PCDに対する製品バイオバーデンの抵抗性の比較をE.2.3 c) の部分サイクルのそれより
短い時間の部分サイクルを用いて以前に評価している及び製品無菌性の試験サンプルから陽
性試験結果がない場合,E.2.3 c) の部分サイクルにばく露した製品試験サンプルの無菌性の
試験を実施する必要性はない。
g) フル滅菌サイクルにばく露した外部PCD(用いる場合)及び内部PCDから試験微生物の生育がない
ことの確認
h) フル滅菌サイクルにばく露した後に,製品機能,安定性及び包装完全性に適合する。
i)
製品が部分及びフル滅菌サイクルの両方にばく露した後に製品EO残留量はJIS T 0993-7の要求事項
に適合していることを確認する。
j)
全ての品質及び規制要求事項に合致する。
注記 この方法から得られる情報及びデータは将来の滅菌プロセスバリデーションの回顧的な補助
に用いることができる。
69
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考文献
[1] JIS Q 9000:2006 品質マネジメントシステム−基本及び用語
注記 対応国際規格:ISO 9000:2005,Quality management systems−Fundamentals and vocabulary(IDT)
[2] JIS Q 9001:2008 品質マネジメントシステム−要求事項
注記 対応国際規格:ISO 9001,Quality management systems−Requirements(IDT)
[3] JIS T 0841-1 最終段階で滅菌される医療機器の包装−第1部:材料,無菌バリアシステム及び包装シ
ステムに関する要求事項
注記 対応国際規格:ISO 11607-1,Packaging for terminally sterilized medical devices−Part 1:
Requirements for materials, sterile barrier systems and packaging systems(IDT)
[4] JIS T 0841-2 最終段階で滅菌される医療機器の包装−第2部:成形,シール及び組立プロセスのバリ
デーション
注記 対応国際規格:ISO 11607-2,Packaging for terminally sterilized medical devices−Part 2: Validation
requirements for forming, sealing and assembly processes(IDT)
[5] JIS Q 14001 環境マネジメントシステム−要求事項及び利用の手引
注記 対応国際規格:ISO 14001,Environmental management systems−Requirements with guidance for
use(IDT)
[6] JIS Q 14040 環境マネジメント−ライフサイクルアセスメント−原則及び枠組み
注記 対応国際規格:ISO 14040,Environmental management−Life cycle assessment−Principles and
framework(IDT)
[7] JIS T 14971 医療機器−リスクマネジメントの医療機器への適用
注記 対応国際規格:ISO 14971,Medical devices−Application of risk management to medical devices
(IDT)
[8] ISO 10993 (all parts),Biological evaluation of medical devices
[9] ISO 14161:2009,Sterilization of health care products−Biological indicators−Guidance for the selection, use
and interpretation of results
[10] ISO 14937:2009,Sterilization of health care products−General requirements for characterization of a
sterilizing agent and the development, validation and routine control of a sterilization process for medical
devices
[11] ISO 15883 (all parts),Washer-disinfectors
[12] ISO 17664,Sterilization of medical devices−Information to be provided by the manufacturer for the
processing of resterilizable medical devices
[13] ISO 22442-1,Medical devices utilizing animal tissues and their derivatives−Part 1: Application of risk
management
[14] ISO 22442-2,Medical devices utilizing animal tissues and their derivatives−Part 2: Controls on sourcing,
collection and handling
[15] ISO 22442-3,Medical devices utilizing animal tissues and their derivatives−Part 3: Validation of the
elimination and/or inactivation of viruses and transmissible spongiform encephalopathy (TSE) agents
[16] TS Z 0032 国際計量計測用語−基本及び一般概念並びに関連用語(VIM)
70
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注記 対応国際規格:ISO/IEC Guide 99:2007,International vocabulary of metrology−Basic and general
concepts and associated terms (VIM)(IDT)
[17] ISO/IEC 90003,Software engineering−Guidelines for the application of ISO 9001:2000 to computer
software
[18] ISO/TS 11139:2006,Sterilization of health care products−Vocabulary
[19] ISO/TS 16775,Packaging for terminally sterilized medical devices−Guidance on the application of ISO
11607-1 and ISO 11607-2
[20] IEC 61010-1,Safety requirements for electrical equipment for measurement, control, and laboratory use−Part
1: General requirements
[21] IEC 61010-2-040,Safety requirements for electrical equipment for measurement, control and laboratory use
−Part 2-040: Particular requirements for sterilizers and washer-disinfectors used to treat medical materials
[22] ANSI/AAMI ST41:1999,Ethylene oxide sterilization in health care facilities: Safety and effectiveness
[23] ANSI/AAMI ST67,Sterilization medical devices−Requirements for products labeled ʻSTERILE. AAMI,
Arlington, VA, 2006
[24] AAMI TIR15,Physical aspects of ethylene oxide. AAMI, Arlington, VA, 2009
[25] AAMI TIR16:2009,Microbiological aspects of ethylene oxide sterilization. AAMI, Arlington, VA, 2009
[26] AAMI TIR28,Product adoption and process equivalence for ethylene oxide sterilization. AAMI, Arlington,
VA, 2009
[27] AS/NZS 4187:2014,Reprocessing of reusable medical devices in health service organizations
[28] EN 556-1,Sterilization of medical devices−Requirements for medical devices to be designated “STERILE”
−Part 1: Requirements for terminally sterilized medical devices
[29] Manufacturers Directive ATEX 94/9/EC, European Parliament and Council, 1994, as amended, 1994
[30] Gillis J., & Schmidt W.C. Scanning electron microscopy of spores on inoculated product surfaces. Medical
Device and Diagnostic Industry. 1983, 5 (6) pp. 46-49
[31] Global Harmonization Taskforce (GHTF) Study Group 1 (SG1), Document No. N29R16:2005 Information
Document Concerning the Definition of the Term “Medical Device”
[32] Holcomb R.G., & Pflug I.J. The Spearman-Karber method of analyzing quantal assay microbial destruction
data. In: Selected Papers on the Microbiology and Engineering of Sterilization Processes, (Pflug I. J. e d.).
Environmental Sterilization Laboratory, Minneapolis, Fifth Edition, 1988, pp. 83-100.
[33] Mosley G.A. Estimating the effects of EtO BIER-Vessel Operating Precision on D-value Calculations, Medical
Device & Diagnostic Industry, April 2002
[34] Mosley G.A., & Gillis J.R. Factors Affecting Tailing in Ethylene Oxide Sterilization. Part 1: When Tailing is an
Artifact and Scientific Deficiencies in ISO 11135 and EN 550. PDA J. Pharm. Sci. Technol. 2004, 58 (2) pp.
81-95
[35] Mosley G. A., G illis J. R., Krushefski G. Evaluating the formulae for integrated lethality in ethylene oxide
sterilization using six different endospore forming strains of bacteria, and comparisons of integrated lethality
for ethylene oxide and steam systems. PDA J. Pharm. Sci. Technol. 2005, 59 (1) pp. 64-86
[36] Mosley G.A., Gillis J.R., Whitbourne J.E. Formulae for Calculations of Integrated Lethality for EtO
Sterilization Processes, Refining the Concepts and Exploring the Applications. Pharm. Tech. 2002, 26 (10) pp.
114-134
71
T 0801:2016 (ISO 11135:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
[37] Pflug I. J. Microbiology and Engineering of Sterilization Processes, Minneapolis. Environmental Sterilization
Services, Minneapolis, Eleventh Edition, 2003
[38] Pflug I.J., Holcomb R. G., Gomez M.M. Thermal Destruction of Microorganisms. In: Disinfection,
Sterilization, and Preservation, (Block S. e d.). Lippincott, Williams & Wilkins, Philadelphia, 2001, pp.
79-129.
[39] Rodriguez A. C., Young B., Caulk K., Zelewski J., Dwasnica S., Aguirre S. Calculating Accumulated Lethality
and Survivorship in EtO Sterilization Processes. MD. 2001 September, p. 1
[40] West K.L. Ethylene oxide sterilization: A study of resistance relationships. In: Sterilization of Medical
Products, (Gaughran E., & Kereluk K. eds.). Johnson & Johnson, New Brunswick, NJ, 1977
[41] Shintani et al. Comparison of D10-value accuracy by the limited Spearman-Karber procedure (LSKP), the
Stumbo-Murphy-Cochran procedure (SMCP), and the survival-curve method (EN). Biomed. Instrum. Technol.
1995, 29 (2) pp. 113-124
[42] Stumbo C.R., Murphy J.R., Cochran J. Nature of Thermal Death Time Curves for P.A. 3679 and Clostridium
Botulinum. Food Technol. 1950, 4 pp. 321-326
[43] USP Monograph on Biological Indicator for Ethylene Oxide Sterilization, Paper Carrier, USP36-NF31 (2013),
page 2659