T 0063:2020 (ISO/IEC Guide 63:2019)
(1)
目 次
ページ
序文 ··································································································································· 1
1 適用範囲························································································································· 1
2 引用規格························································································································· 1
3 用語及び定義··················································································································· 2
4 “安全”,“安全な”,“有効な”及び“有効性”という用語の使用 ·············································· 5
4.1 安全 ···························································································································· 5
4.2 安全な ························································································································· 5
4.3 有効な ························································································································· 5
4.4 有効性 ························································································································· 6
5 医療機器規格に安全側面を導入するための原則 ······································································ 6
5.1 安全側面を導入した医療機器規格の適用範囲 ······································································ 6
5.2 安全側面を導入した医療機器規格の目的 ············································································ 6
5.3 規格のタイプ ················································································································ 7
5.4 安全の実用面の考慮 ······································································································· 8
5.5 医療機器規格の調整 ······································································································· 8
5.6 規格の規制上又は法令上の使用の意味合い ········································································· 8
6 リスクの性質··················································································································· 9
6.1 リスクの要素 ················································································································ 9
6.2 リスクの系統的又は偶発的性質 ························································································ 9
7 安全側面を導入した医療機器規格の開発のためのリスクに基づくプロセス ································· 11
7.1 一般 ··························································································································· 11
7.2 予備作業 ····················································································································· 11
7.3 原案作成 ····················································································································· 14
7.4 規格のバリデーション ··································································································· 23
7.5 結論 ··························································································································· 23
8 リスクマネジメントの枠組みにおける安全側面を導入した医療機器規格の適用の概要 ·················· 23
附属書A(参考)製品安全規格及びプロセス安全規格 ································································ 25
附属書B(参考)リスク情報 ································································································· 26
参考文献 ···························································································································· 27
T 0063:2020 (ISO/IEC Guide 63:2019)
(2)
まえがき
この規格は,産業標準化法第12条第1項の規定に基づき,一般社団法人日本医療機器産業連合会
(JFMDA)から,産業標準原案を添えて日本産業規格を制定すべきとの申出があり,日本産業標準調査
会の審議を経て,厚生労働大臣及び経済産業大臣が制定した日本産業規格である。
この規格は,著作権法で保護対象となっている著作物である。
この規格の一部が,特許権,出願公開後の特許出願又は実用新案権に抵触する可能性があることに注意
を喚起する。厚生労働大臣,経済産業大臣及び日本産業標準調査会は,このような特許権,出願公開後の
特許出願及び実用新案権に関わる確認について,責任はもたない。
日本産業規格 JIS
T 0063:2020
(ISO/IEC Guide 63:2019)
医療機器規格における安全側面の
開発及び導入の指針
Guide to the development and inclusion of aspects of
safety in standards for medical devices
序文
この規格は,2019年に第3版として発行されたISO/IEC Guide 63を基に,技術的内容及び構成を変更
することなく作成した日本産業規格である。
この規格は,規格作成者に対し,医療機器規格の開発において安全側面をどのように導入するかについ
ての実践指針を提供する。対象とする医療機器規格は,医療機器のリスクマネジメント全般を規定するJIS
T 14971のほかに,特定のリスクを評価しコントロールするための特別な方法及び要求事項を規定する特
定の規格を含む。この規格は,医療機器分野のニーズに対応するために,一般的な安全側面の導入指針で
あるJIS Z 8051:2015及びリスクマネジメントの原則に基づいている。
この規格で点線の下線を施してある参考事項は,対応国際規格にはない事項である。
この規格の箇条3の定義の中の太字は,この規格の箇条3で定義した用語である。
1
適用範囲
この規格は,確立されたリスクマネジメントの概念及び方法論に基づいて,医療機器規格の作成者が規
格に安全側面を導入する際の要求事項及び推奨事項について規定する。
この規格は,人,財産,環境又はそれらの組合せの安全に関する全ての側面に対して適用する。
この規格において,“製品”という用語は,医療機器に加えて,複数の医療機器で構成されるシステムを
含む。システムは,場合によっては非医療機器とも組み合わされる。
注記 この規格の対応国際規格及びその対応の程度を表す記号を,次に示す。
ISO/IEC Guide 63:2019,Guide to the development and inclusion of aspects of safety in International
Standards for medical devices(IDT)
なお,対応の程度を表す記号“IDT”は,ISO/IEC Guide 21-1に基づき,“一致している”こと
を示す。
2
引用規格
この規格には,引用規格はない。
2
T 0063:2020 (ISO/IEC Guide 63:2019)
3
用語及び定義
この規格で用いる主な用語及び定義は,次による。
3.1
危害(harm)
人の受ける傷害若しくは健康障害,又は財産若しくは環境の受ける害
(出典:JIS Z 8051:2015の3.1を変更)
3.2
ハザード(hazard)
危害(3.1)の潜在的な源
(出典:JIS Z 8051:2015の3.2)
3.3
危険状態(hazardous situation)
人,財産又は環境が,一つ以上のハザード(3.2)にさらされる状況
(出典:JIS Z 8051:2015の3.4)
3.4
意図する使用(intended use)
製造業者(3.6)が提供する仕様,取扱説明及び情報で意図している,製品,プロセス又はサービスの
使用
注釈1 意図する医学的適応,患者集団,相互に作用し合う対象の体の部分又は生体組織の種類,ユー
ザープロファイル,使用環境及び動作原理が,意図する使用の典型的要素である。
3.5
ライフサイクル(life cycle)
医療機器(3.7)の初期構想から最終的な使用停止及び廃棄に至るまでの一連の全ての段階
3.6
製造業者(manufacturer)
医療機器(3.7)の設計及び/又は製造が自分自身によるか,又は他の人による行為かにかかわらず,
その名の下に,使用に供するために医療機器(3.7)を作ることを意図し,医療機器(3.7)の設計及び/
又は製造に責任をもつ自然人又は法人
注釈1 この“自然人又は法人”は,利用可能とする又は販売することを意図した国又は法的管轄にお
いて,法的管轄で規制当局によって他の人に特別に責任を負わす場合を除き,適用される全て
の医療機器の規制要求事項に適合させる最終的な法的責任をもっている。
注釈2 製造業者の責任は,他のGHTF指針文書に記載されている。これらの責任には,市販前要求事
項並びに有害事象報告及び是正措置の通知のような市販後要求事項の両方に適合することを含
んでいる。
注釈3 “設計及び/又は製造”は,仕様開発,生産,成型加工,組立,加工,包装,再包装,ラベリ
ング,再ラベリング,滅菌,据付け若しくは医療機器の再製造,又は医療目的のために利用可
能な他の製品及び機器を一緒に収集してまとめることを含む場合がある。
注釈4 個々の患者に対して他の人が既に供給した医療機器を,取扱説明書に従って組立又は調整する
人は,製造業者ではない。ただし,指定された組立又は調整は,医療機器の意図する使用を変
3
T 0063:2020 (ISO/IEC Guide 63:2019)
更しないことが前提である。
注釈5 医療機器の元々の製造業者の代理としてではなく,医療機器の意図する使用を変更する人,医
療機器を改造する人,又は自身の名の下にそのような医療機器を利用可能にする人は,変更し
た医療機器の製造業者とみなされる。
注釈6 既存のラベルを覆ったり,変更したりすることなく,医療機器又はその包装に自身の所在地及
び連絡先だけを表示する指定代理人,ディストリビューター及び輸入業者は,製造業者とはみ
なさない。
注釈7 附属品は医療機器の規制要求事項の対象となるため,その附属品の設計及び/又は製造に関し
て責任をもつ者は,製造業者とみなす。
(出典:GHTF/SG1/N055:2009の5.1のNOTE 3を変更し,“を含めてよい”を,“を含む場合がある”
に置換え)
3.7
医療機器(medical device)
計器,器械,用具,機械,器具,植込み用具,体外診断薬,ソフトウェア,材料又はその他の同類のも
の若しくは関連する物質であって,単独使用か又は組合せ使用かを問わず,製造業者(3.6)が人体への
使用を意図し,その使用目的が次の一つ以上であり,
− 疾病の診断,予防,監視,治療又は緩和,
− 負傷の診断,監視,治療,緩和又は補助,
− 解剖学的又は生理学的なプロセスの検査,代替,修復又は支援,
− 生命支援又は維持,
− 受胎調整,
− 医療機器(3.7)の消毒,
− 人体から採取される検体の体外試験法による情報提供,
さらに,薬学,免疫学又は新陳代謝の手段によって,体内又は体表において意図するその主機能を達成す
ることはないが,それらの手段によって意図する機能の実現が補助される場合があるもの
注釈1 法的管轄によって医療機器に該当するか否かが分かれる場合がある製品には,次のものがある。
− 消毒剤
− 身体障害者用の補助器具
− 動物及び/又はヒト組織に由来する機器
− 体外受精又は生殖補助技術用の機器
(出典:GHTF/SG1/N071:2012の5.1を変更し,“補助されてもよいもの”を,“補助される場合がある
もの”に置換え,また,NOTEを変更し,“否かが分かれる”を,“否かが分かれる場合がある”に置換
え)
3.8
合理的に予見可能な誤使用(reasonably foreseeable misuse)
容易に予測可能な人間の行動によって引き起こされる使用であるが,製造業者(3.6)が意図しない方
法による製品又はシステムの使用
注釈1 容易に予測可能な人間の行動は,様々なタイプのユーザー(例えば,一般の人及び専門家)の
行動を含む。
注釈2 合理的に予見可能な誤使用は,意図的である場合も意図的でない場合もある。
4
T 0063:2020 (ISO/IEC Guide 63:2019)
(出典:JIS Z 8051:2015の3.7を変更し,“供給者”を“製造業者”に置換え,また,注記1の例を変
更し,注記2を削除し注釈2を追加)
3.9
残留リスク(residual risk)
リスクコントロール(3.12)手段を実施した後にも残るリスク(3.10)
(出典:JIS Z 8051:2015の3.8を変更し,“リスク低減方策”を“リスクコントロール手段”に置換
え)
3.10
リスク(risk)
危害(3.1)の発生確率とその危害(3.1)の重大さ(3.17)との組合せ
注釈1 発生確率には,危険状態への暴露及び危害の回避又は制限の可能性を含む。
(出典:JIS Z 8051:2015の3.9を変更し,“危険事象の発生”を注釈1から削除)
3.11
リスク分析(risk analysis)
ハザード(3.2)を特定するための及びリスク(3.10)を推定するための利用可能な情報の体系的な使
用
(出典:JIS Z 8051:2015の3.10を変更)
3.12
リスクコントロール(risk control)
規定したレベルまでリスク(3.10)を低減するか又はそのレベルでリスク(3.10)を維持するという決
定に到達し,かつ,そのための手段を実施するプロセス
3.13
リスク推定(risk estimation)
危害(3.1)の発生確率とその危害(3.1)の重大さ(3.17)に対して重み付けをするために用いるプロ
セス
3.14
リスク評価(risk evaluation)
判断基準に照らして推定したリスク(3.10)の受容可能性を判断するプロセス
3.15
リスクマネジメント(risk management)
リスク(3.10)の分析,評価,コントロール及び監視に対する,マネジメント方針,手順及び実施の体
系的な適用
3.16
安全(safety)
受容できないリスク(3.10)がないこと
3.17
重大さ(severity)
ハザード(3.2)から生じる可能性がある結果の尺度
5
T 0063:2020 (ISO/IEC Guide 63:2019)
3.18
最新の技術水準(state of the art)
ある時点での,科学,技術及び経験を結集した知見に基づいた,製品,プロセス及びサービスに関する
技術力の到達段階
注釈1 最新の技術水準は,技術及び医学の優れた実践として現在一般に受け入れられているものを具
体化したものである。最新の技術水準は,必ずしも技術的に最も進んだ解決策を意味しない。
最新の技術水準は,この規格では“一般に認められた最新の技術水準”として規定する場合が
ある。
(出典:JIS Z 8002:2006の1.4を変更し,注釈1を追加)
3.19
検証(verification)
客観的証拠を提示することによって,規定要求事項が満たされていることを確認すること
注釈1 検証のために必要な客観的証拠は,検査の結果,又は別の方法での計算の実施若しくは文書の
レビューのような他の形の確定の結果であることがある。
注釈2 検証のために行われる活動は,適格性確認プロセスと呼ばれることがある。
注釈3 “検証済み”という言葉は,検証が済んでいる状態を示すために用いられる。
(出典:JIS Q 9000:2015の3.8.12)
4
“安全”,“安全な”,“有効な”及び“有効性”という用語の使用
4.1
安全
医療機器規格における“安全”という用語は,説明的な形容詞として使用するよりはむしろ名詞として
使用することが望ましい。形容詞として使用すると,リスクがないことを保証していると誤解されやすい
ので,“安全”という用語は,可能な限り目的を示す用語に置き換えることが望ましい。
例 “安全ヘルメット”の代わりに“保護ヘルメット”,“安全インピーダンス装置”の代わりに“保護
インピーダンス装置”とする。
4.2
安全な
医療機器規格における“安全な”という用語は,認識された危険状態に由来するリスクが受容可能なレ
ベルまで低減されている状態を示すために使用することが望ましい。
“安全な”という用語は,認識された危険状態に由来するリスクが受容可能なレベルまで低減されてい
る状態と,製品が意図する使用を達成している状態との間でのバランスがとれた状況を記載するために,
“有効な”という用語だけとともに使用することが望ましい。その他の形で“安全な”という用語を使用
している場合は,可能な限り,目的を示す用語に置き換えることが望ましい。
例 “安全な床仕上げ材”の代わりに“滑り止め処理床仕上げ材”とする。
4.3
有効な
医療機器規格における“有効な”という用語は,医療機器がその意図する使用を満たしていることを示
すために使用することが望ましい。
6
T 0063:2020 (ISO/IEC Guide 63:2019)
4.4
有効性
“有効性”という用語は,医療機器が使用される文脈に依存する関連概念の多様性を表すために,医療
機器規格において使用することが可能である。規格作成者は,この規格に定める文脈と異なる場合,その
規格の文脈内の意味を慎重に定めた上で,一貫性をもってそれを使用する必要がある。この規格では,こ
の用語を,リスクコントロール手段の検証という文脈で使用する。
例 JIS T 62366-1:2019の3.4においては,“有効性”を,“ユーザーが,指定された目標を達成する上で
の正確さ及び完全さ”と定義している。IEC 80001-1:2010の2.6においては,“有効性”を“患者及
び責任部門のために意図する結果を生み出す能力”と定義している。
5
医療機器規格に安全側面を導入するための原則
5.1
安全側面を導入した医療機器規格の適用範囲
安全側面を導入した医療機器規格の計画及び開発には,製造業者,ユーザー,規制当局,その他の利害
関係者を含めた包括的アプローチが求められる。この規格は,様々な医療機器規格を担当する委員会が,
規格を作成する際に安全の取扱いに対する首尾一貫したアプローチを行うことを支援しようとするもので
ある。それらの規格の適用範囲を定義することによって,各規格は特定の側面に制限され,また,各規格
が他の全ての関連する側面に対してより広範に適用できる規格を引用することが確実になる。役に立つ規
格体系は,次のように構築される。
− 基本規格:全ての種類又は幅広い製品,プロセス及びサービスに適用する一般的安全側面に関する基
本概念,原則及び要求事項を含むもの(基本規格は,しばしば,水平規格とも称される。)
− グループ規格:二つ以上の専門委員会又は分科委員会が扱う複数の製品,プロセス又はサービスに適
用する安全側面を含むもの。できる限り基本規格を引用する。
− 特定の製品(のファミリー)規格及び/又はプロセス規格:単独の専門委員会又は分科委員会の担当
範囲内の個別製品若しくはファミリー製品,プロセス又はサービスに適用する,全ての必要な安全側
面を含むもの。できる限り基本規格及びグループ規格を引用する(製品規格及びプロセス規格は,し
ばしば,垂直規格とも称される。)。
− 安全側面を含むその他の規格:ただし,安全側面だけを取り扱うものではなく,できる限り基本規格,
グループ規格,並びに製品規格及びプロセス規格を引用したもの
上記の規格体系は,JIS Z 8051:2015の7.1に基づいている。
医療機器の安全側面を取り扱う要求事項は,上記の各階層に分類可能な様々なタイプの規格(5.3参照)
に含めることが可能である。
5.2
安全側面を導入した医療機器規格の目的
安全に関する側面を導入した医療機器規格の目標は,予測可能な一貫した安全レベルをもつ医療機器の
開発及び製造を支援することである。
この目標を達成するために,これらの規格は,次のとおりであることが望ましい。
a) 安全な及び有効な医療機器の設計及び製造において,製造業者を支援する。
注記 用語“安全な”及び“有効な”の使用の指針については,4.2及び4.3を参照。
b) 法的及び市場要求事項への適合を評価することにおいて,製造業者,認証機関,試験機関又は試験所,
7
T 0063:2020 (ISO/IEC Guide 63:2019)
及び規制当局を支援する。
c) 医療機器の使用に関連するリスクをマネジメントすることにおいて,ヘルスケアプロバイダー(例え
ば,医療従事者,医療機関)及びユーザーを支援する。
上記の利害関係者の支援に適する安全側面を導入した医療機器規格を作成するため,規格作成者は,こ
の規格のリスクに基づく枠組みを採用することが望ましい。
安全側面を含む医療機器規格を原案作成する場合,規格作成者は,この目標を支援し,規格が正確に適
用されることを確実にするために,箇条3の定義を慎重に順守することが望ましい。
5.3
規格のタイプ
5.3.1
製品規格
製品規格には,例えば,次がある。
a) 安全又は性能についてのパラメーターを規定し,これらのパラメーターへの適合を実証するために使
用することができる標準試験方法を含む規格
b) 安全に関する情報の提供を要求する規格,又は安全及び性能に対して宣言された合否基準の順守が必
要な場合の試験方法規格
製品規格が安全な及び有効な医療機器にどのように寄与できるかについては,A.1を参照。
5.3.2
プロセス規格
プロセス規格には,例えば,次がある。
a) 製造業者が一貫して仕様を満たす医療機器を設計,開発及び生産することを可能とする枠組みについ
て定めた品質マネジメント又はリスクマネジメントなどのマネジメントシステム規格
b) 製造業者が一貫して安全な及び有効な医療機器を設計,開発及び生産することが可能となる枠組みに
ついて定めたプロセス規格(例えば,滅菌,生物学的評価,臨床調査)
プロセス規格が安全な及び有効な医療機器にどのように寄与できるかの検討については,A.2を参照。
規格のタイプによっては,製品規格及びプロセス規格の特質を兼ね備えているため,これらのカテゴリ
ーの一つに容易に割り当てることができないものがある。例を,5.3.3及び5.3.4に示す。
5.3.3
据付け規格及び環境規格
据付け規格及び環境規格は,複雑な,統合システム,能動医療機器及び情報技術(IT)環境で動作する
医療機器に対して一般に適したものである。据付け規格及び環境規格には,例えば,次がある。
a) 構造及び据付け規格(例えば,X線遮蔽,電気配線)
b) 複数の機器を相互接続して単一システムとするための注意事項及び手順を規定するシステム規格
c) 恒久的に据え付けた装置及びシステムの初回の使用前に行う試験及び検査手順を規定する試運転のた
めの規格
d) 医療機器がその環境に悪影響を与えないこと,及び環境が医療機器の性能を劣化又は損なうことがな
いことを確実にするための,注意事項及び試験を規定する環境規格[例えば,電磁両立性(EMC)規
格]
e) ITセキュリティ規格又はサイバーセキュリティ規格
8
T 0063:2020 (ISO/IEC Guide 63:2019)
5.3.4
運用規格
運用規格は,例えば,次がある。
a) 医療機器の安全及び有効性が機器の耐用年数にわたって維持されることを確実にするための,運用中
定期試験規格
b) 安全に関する医療機器の適切な機能及び正確さを継続して確実にするための,品質保証及び校正規格
5.4
安全の実用面の考慮
リスクは,製品,プロセス又はサービスに関するその他の要求とバランスをとる必要がある。これらの
要求には,ベネフィット,適合性及び利用可能性が含まれる。規格作成者は,製造業者の必要な労力(例
えば,必要な文書化又は試験)のレベルをリスクのレベルに合わせることに留意することが望ましい。
ゼロリスクは達成不可能であるため,安全は受容できないリスクがないことと定義される。実用的なア
プローチは,一般に認められた最新の技術水準及び既知の利害関係者の懸念などの入手可能な情報を考慮
した,高いレベルの安全及び健康の保護がもたらされる受容可能なリスクのレベルを確立することである。
医療機器の安全の評価において,一部の医療機器は,その操作手段,構成又は使用環境の理由から,有
効性を低下させることなしには排除できない,固有のリスクがあることも考慮する必要がある(例えば,
外科用レーザー機器,電気外科機器,X線画像機器,放射線治療機器)。
世界の様々な地域には,医療機器の安全に関する判断を含めて,医療及び健康に関する習慣上の違いが
存在する。さらに,技術的及び社会的価値は変化するため,安全であるとみなされるものは時間とともに
進化する。これらの課題は,しばしば,技術的要求事項を適用する個別の条件を特定することによって対
処し得る。
5.5
医療機器規格の調整
それぞれの新しい医療機器規格の開発は,既存の医療機器及び規格だけでなく,国,地域及び国際的法
令の関係において検討する必要がある。新しい規格は,関連のある限り,利便性又は明瞭さによって正当
な理由付けがされる場合,本文の参照又は複製のいずれかによって,既存規格の本体を活用することが望
ましい。
5.6
規格の規制上又は法令上の使用の意味合い
規格作成者は,自らが開発する規格について,考えられる法令上及び規制上の意味合いを認識すること
が望ましい。
安全な及び有効な医療機器は,多くの国々で販売及び使用が規制されているが,それらの国々の規制当
局にとっては特別な関心事である。しかし,規格は,特定の規制だけに対処するような規格作成をしない
ことが望ましい。
規格は規制及び法律において引用されることがあり,その場合,規格それ自体が法的拘束力のあるもの
となる。また,特定の規格に適合する医療機器を規制に“適合するとみなす”規制スキームもある。
規格作成者は,規格の適用が規制当局によって変更される場合もあることを認識する必要がある。
規格は,訴訟において,社会が期待するものとして引用されることもあるので,社会の期待に対して適
合するために使用可能である。
9
T 0063:2020 (ISO/IEC Guide 63:2019)
経験から,参考附属書又は論理的根拠及び例をもつ注記などの非規定情報が,規定として誤解される可
能性があることが示されている。論理的根拠を説明する以外の参考附属書の導入は慎重に検討し,適切に
表現することが望ましい。
6
リスクの性質
6.1
リスクの要素
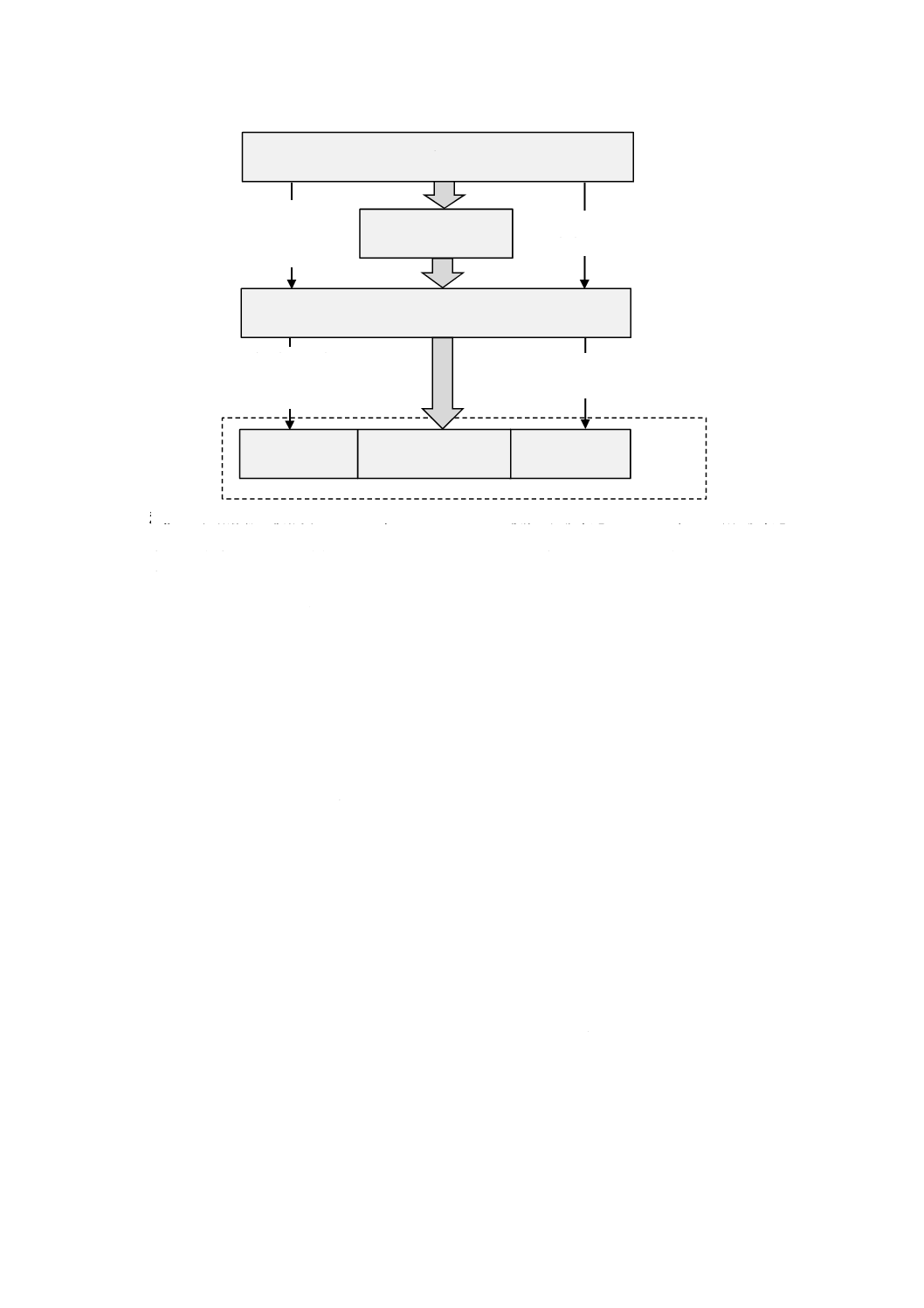
特定の危険状態に関連するリスクは,次の要素の組合せである(図1参照)。
a) 危害の発生確率:危害の発生確率は,次の組合せであると考えられる。
− 事象の特定の順序又は組合せが危険状態に至る(すなわち,ハザードへの暴露)確率P1
− 危険状態が危害に至る確率P2
b) 危害の重大さ:危害は,危険状態の結果,起こり得る。
医療機器の複雑さ,意図する使用,又は暴露の頻度若しくは期間に応じて,危害の発生確率は,二つの
確率(P1,P2)の組合せとして又は一つの確率(P)として表すことが可能である。図1に,これらの要素
がどのように相互に関連しているかを示す。P1及びP2への分解は,必須ではない。
次に,それぞれ個別に推定した危害の重大さ及び発生確率を組み合わせて,リスクを評価する。
危害の発生確率を推定することができない場合,通常は,危害の重大さだけに基づいてリスクを評価す
る必要がある。危害の重大さが大きく,リスクコントロール手段の有効性が低くなるほど,関連するリス
クコントロールに要求される厳密さは高くなる。
P1又はP2のいずれかを推定することができて,他方の確率を推定することができない状況では,未知の
確率を1にした慎重なアプローチを行うことが可能である。この場合は,リスクを危害の重大さ及び発生
確率の慎重な推定値に基づいて評価することが可能である。
注記 附属書Bに,リスク情報を入手するための情報源及び方法の使用に関する指針を示す。
6.2
リスクの系統的又は偶発的性質
6.2.1
リスクの原因のタイプ
リスクは,系統的な又は偶発的な原因のいずれかから発生し得る。その結果,危害の発生確率は,系統
的な又は偶発的な原因のいずれかと関連付けることが可能である。
例えば,製造プロセス内の系統的欠陥(不十分な洗浄工程,滅菌後の不十分なエアレーションなど)に
起因して,有毒物質が製品に含まれる場合がある。一方,原材料のランダムな変動又はプロセスのランダ
ムな変動が,有毒物質の存在につながる場合もある。危害の発生確率の定量的推定を正確かつ信頼性のあ
るデータに基づいて実施できる場合,又は合理的な定性的推定が可能な場合は,リスク推定の信頼性は向
上する。しかし,これは常に達成可能なわけではない。危害の発生確率の推定の正確さに疑義がある場合
は,多くの場合,危害の発生確率について幅広い範囲を定めるか,又はある特定の値よりも悪くないと決
定する必要がある。
規格作成者は,自らが作成する規格について,系統的な原因と偶発的な原因との間の違いを考慮する必
要がある。
10
T 0063:2020 (ISO/IEC Guide 63:2019)
注記1 医療機器の複雑度によっては,一つのハザードが複数の危険状態に至ったり,その各危険状態
が複数の危害に至ったりすることがある。
注記2 危害の発生確率(P)は,独立した確率P1及びP2の組合せとすることが可能である。
注記3 細い矢印はリスク分析の要素を表し,太い矢印はハザードがどのように危害に至るかを示す。
図1−ハザード,一連の事象,危険状態及び危害の関係の図解例
6.2.2
系統的な原因から発生するリスク
あらゆる活動における誤りは系統的な原因を誘引することがあり,これが,インプット又は条件の特定
の組合せが発生したときに故障を生じさせる。こうした誤りは,製品のライフサイクルのいかなる時点で
も発生し得る。系統的な原因が発生する確率は,推定することが困難な場合もある。
例を次に示す。
a) ソフトウェアを原因とする故障
b) 不適切な設計又は取扱説明書の欠陥
c) 新しい医学的適応
系統的な原因によって危険状態が発生する場合,系統的な原因が発生する確率は,危害の発生確率と同
一ではない。原因は必ずしも危険状態をもたらすわけではなく,また,危険状態が必ずしも危害をもたら
すわけではない。
系統的な原因から発生するリスクに適用できるリスクコントロール手段の例には,次が含まれる。
− 設計,開発及び製造に適用するプロセスの厳格さ:通常,設計,開発又は製造に使用するプロセスが
厳格になるほど,発生する又は検出されないまま残る系統的な故障の確率は低くなる。
− リスクコントロール手段の冗長性を適用する:複数の独立したリスクコントロール手段は,通常,特
定のリスクに対する全体的保護への信頼性を高める。
− 危険状態及びその後の危害に関与する,二つ以上の個別の事象が発生する期間を縮小する:具体的に
は,定期的自主点検から定期的保守までの期間を短縮する。
− 重要なパラメーターの継続的監視,並びにその後の評価及び是正処置のプロセス及びメカニズムを適
リスク
ハザード
危険状態
一連の事象
危険状態が
起こる確率
(P1)
危険状態が危害に
至る確率
(P2)
重大さに影響を
与える状況
危害の発生確率
(P=P1×P2)
危害の重大さ
危害
重大さに影響を
与える状況
11
T 0063:2020 (ISO/IEC Guide 63:2019)
用する。
6.2.3
偶発的な原因から発生するリスク
多くの事象については,故障が発生する確率に数値を与えることが可能である。ハザード並びに発生す
る危険状態の確率(ハザードへの暴露)(図1のP1)及び危害に至る危険状態の確率(図1のP2)に影響
を与える状況に関して十分な情報を得ている場合だけ,定量的推定を適用することが可能である。
偶発的な故障の幾つかの例を,次に示す。
− 電子的組立品内の集積回路など,一つの部品の故障の原因となる電力サージ
− 劣化の原因となる体外診断用(IVD)試薬の汚染
− 生物学的反応に至る医療機器内又は医療機器上の感染因子の存在
− アレルギー反応に至る医療機器内又は医療機器上の有毒物質の存在
7
安全側面を導入した医療機器規格の開発のためのリスクに基づくプロセス
7.1
一般
安全側面を導入した医療機器規格を作成する場合,規格作成者は,リスクの受容可能性の判断基準の決
定,リスク分析,リスク評価,リスクコントロール,全体的な残留リスクの受容可能性の評価を含む,リ
スクマネジメントの計画で構成するリスクに基づく枠組みを使用することが望ましい。この箇条の情報は,
製品規格及びプロセス規格の両方に適用可能である。
以降の細分箇条では,従うべき手順上のステップを記載し,規格を開発するときに使用するリスクマネ
ジメントの枠組みについて記載する。
7.2
予備作業
7.2.1
安全側面を導入した規格の制定又は改正の必要性の特定
安全側面を導入した規格を作成又は改正するプロジェクトに着手する前に,プロジェクトの提案者は,
規格で取り扱うべき内容,及び意図する読者は誰かを特定することが望ましい。これは,通常,次の質問
に答えることによって達成される。
a) 規格は,どの医療機器又は医療機器関連プロセスに適用するか
b) 既に規格はあるか
c) 規格の対象者は誰か
− 誰が,どのように規格を適用するか
− 起こり得る環境への影響を含め,誰が及び/又は何が規格の影響を受けるか
− 規格を適用する人及び/又は規格に影響される人は,そこに何を求めるか
− 規格の開発期間中,どの利害関係者が関与することが必要か
d) どのタイプの規格になるか
− 基本規格
− グループ規格
− 特定の製品又はプロセス規格
12
T 0063:2020 (ISO/IEC Guide 63:2019)
e) 規格の目的は何か
− 取り扱う安全に影響し得る特質を特定するか
− 規格は,試験のために使用されるか
− 規格は,適合性評価の基礎として役立つか
− 規格は,規格の発行前に市販された製品(すなわち,レガシー製品)の適合性を評価するために使
用されるか
この情報は,担当専門委員会への新規作業項目提案に含めることが可能である。
7.2.2
規格開発におけるリスクマネジメントの枠組みの確立
規格に関するリスクマネジメント作業は,対象とする安全側面の特定から始まる。この段階では,全て
の関連情報(例えば,事故データ,調査報告書)を収集することが不可欠である。次に,規格の基礎とし
て役立つ詳細なアウトラインを作成することが望ましい。規格を開発するために必要な専門知識を,委員
会内部に集める必要がある。このような知識には,例えば,次が含まれる。
− 製品又はプロセスの詳細な実務知識
− 様々な情報源からの要求事項及びガイドラインで,開発する規格に関する一般的なものと固有のもの
の両方
− 人間の行動研究及び人体測定データ
− 欠陥による傷害及び/又はインシデントのデータ,並びに製品のリコール履歴
− 製品の健康及び環境への潜在的な影響についての知識
− 製品の最終ユーザーの経験に基づくフィードバック
− 潜在的リスクコントロール手段(保護手段)についての知識
− 今後の製品開発の動向についての知識
− 業界標準及びガイドライン
− 関連する利害関係者からの利用可能な専門知識及び科学的な助言
− 法的及び規制上の要求事項
規格の基本的な内容が決まったら,次の安全側面を考慮することが望ましい(必ずしも,これらの全て
が所与の規格に関連するわけではない。)。
− 意図する使用及び合理的に予見可能な誤使用
− 期待する使用条件下で発揮される製品の能力
− 環境適合性(例えば,電磁的,機械的及び気候的現象の考慮)
− 人間工学的要因及びユーザビリティ的要因
− 既存の関連規格
− 既知のリスクコントロール手段の利用可能性及び/又は信頼性
− 保守性(“サービス保守”を含む。例えば,サービス可能な品目へのアクセスのしやすさ,及び燃料補
給又は注油の方法)
− 保守及びケア
− 既知のリスクコントロール手段の永続性及び依存性
− 廃棄可能性(関連する指示書を含む。)
13
T 0063:2020 (ISO/IEC Guide 63:2019)
− 製品の最終ユーザーの特別なニーズ(例えば,潜在的ニーズ,顕在的ニーズ)
− 故障特性
− マーキング,情報及びラベリング
− 組立説明書
− 安全に関する情報
7.2.3
リスクの受容可能性の判断基準
7.2.3.1
リスクの判断基準の確立
規格作成者にとっては,規格の要求事項が適切かどうかを評価するのに適したリスクの判断基準を確立
することが重要である。
まず,規格作成者は,リスクの重大性を評価するために使用する判断基準を定義することが望ましい。
この判断基準は,技術的能力及びヘルスケアの提供価値,目的及び資源を反映することが望ましい。一部
の基準は,法律上及び規制上の要求事項並びに対象とする技術に適用するその他の要求事項によって課さ
れることがあったり,又はそれを由来としたりすることがある。患者,介護者及びその他の人については,
異なるリスクの受容可能性の判断基準を定めることが可能である。
リスクの判断基準を定めるとき考慮する要素には,次のことを含めることが望ましい。
− 技術に伴って発生することがある原因及び結果の性質及びタイプ,並びにそれをどのように測定する
か
− 確率をどのように定義するか
− 確率及び/又は危害若しくは結果が関連する期間
− リスクレベルをどのように決定するのがよいか
− 利害関係者の意見をどのように判断するのがよいか
7.2.3.2
受容可能なリスクの決定
リスクの判断基準を使用して,規格作成者は,どのようなリスクレベルが受容可能とみなすことができ
るかを決定する。受容可能なリスクを決定する方法には次が含まれるが,これらだけに限らない。
− 一般に認められた最新の技術水準を表し,臨床リスクとベネフィットとの比に対してバランスがとれ
た受容可能なリスクの実証に対する要求事項を含む,適用できる基本規格及びグループ規格の使用
− 一般に認められた最新の技術水準とみなされている,既に使用に供されている医療機器から明らかに
なっているリスクレベルの比較
− 専門家の意見の使用
− 臨床データを含めた科学的調査結果の使用
− 利害関係者の関心事及び社会的期待
− 適用される国又は地域の規制
7.2.3.3
最新の技術水準
リスクの判断基準を定めるとき,規格作成者は,技術及び医学において現在,一般によい慣行として受
け入れられているものを具体化した,一般に認められた最新の技術水準を考慮することが望ましい。最新
の技術水準とは,必ずしも,技術的に最も進んだ解決策を意味するわけではない。
14
T 0063:2020 (ISO/IEC Guide 63:2019)
7.2.3.4
利害関係者の関心事
リスクの認知が,しばしば,経験的に求めたリスク推定と異なることはよく知られている。したがって,
どのようなリスクが受容可能であるかを決定するときは,広く様々な利害関係者がリスクをどう認知する
かを考慮することが望ましい。世論の期待を満たすためには,一部のリスクに重要度を付加することが必
要なことがある。場合によっては,特定した利害関係者の関心事が社会の価値を反映しており,これらの
関心事が考慮されているとみなすことが唯一の選択肢であることがある。
7.2.3.5
社会的期待
リスクの認知は社会ごとに異なり得るものであり,このことを考慮に入れることが望ましい。それにも
かかわらず,安全側面を導入した医療機器規格は,承認プロセスによって受け入れられた場合には,異な
る社会的期待が含まれるものと想定される。
7.2.3.6
確率が推定できないときのリスクの決定及び受容可能性
危険状態に至る一連の事象の確率(6.1に定義したP1)又は危害に至る危険状態の確率(同じく6.1に定
義したP2)のいずれかが推定できない状況については,未知の確率を1として,危害の発生確率を慎重に
推定することが可能である。危害の発生確率を全く推定できないときは,生じる可能性のある結果を考慮
してリスクを決定し,リスクの受容可能性を評価することが望ましい。
7.3
原案作成
7.3.1
一般
次に示す規則及び推奨事項は,JIS Z 8301を補足し,安全に関連する側面を導入した規格になることを
意図する規格の原案作成に適用する。
安全に関連する側面を導入した医療機器規格の作成者は,規格の主題である製品又はプロセスに関連す
るハザード及び危険状態に精通していることが望ましい。製品規格の場合,作成者は,(例えば,附属書の
形式で)特定の医療機器又はシステムに共通する既知のハザード及び/又は危険状態のリストを含めるこ
とを考慮することが望ましい。プロセス規格の場合,作成者は,規格の適用がどのようにリスクを低減す
るかを説明する論理的根拠を含めることが望ましい。
可能な限り,規格は,ハザードの除去又はリスク低減において重要な要求事項を含むことが望ましい。
これらの要求事項は,リスクコントロール手段(例えば,保護手段)の観点から表すことが望ましい。リ
スクコントロール手段は,検証が可能であるように規格で規定することが望ましい。
リスクコントロール手段の要求事項は,次のようであることが望ましい。
a) 正確かつ明確に理解可能な文章で規定する
b) 技術的に正しい
c) 検証可能である
規格は,要求事項が満たされていることを検証する方法について明確で完全な規定を含むことが望まし
い。
パフォーマンスに基づくリスクコントロール手段を規格で規定する場合,要求事項には,次を含めるこ
とが望ましい。
15
T 0063:2020 (ISO/IEC Guide 63:2019)
− コントロールの対象となるリスクのリスト
− 各リスクコントロール手段の明確なパフォーマンス要求事項
− パフォーマンス要求事項への適合を判定するための詳細な検証方法
リスクコントロールの要求事項は,ただ単に設計特性を説明するだけではなく,パフォーマンス特性(パ
ラメーター)及びその値を用いて,安全に関する検証可能なパフォーマンスとして表現する(例えば,ブ
レーキングシステムに要求されるパフォーマンス特性として,y km/hで走行する自動車に必要な制動距離
x m)ことが望ましい。
また,規格で定義されていない限り,主観的な用語又は主観的な言葉の使用は最小限にすることが望ま
しい。
注記 対応国際規格のNOTE 1及びNOTE 2は推奨事項のため,本文に移動した。
規格作成者は,規格開発の簡単な経緯又は決定の論理的根拠の作成を考慮することが望ましい。
7.3.2
リスクマネジメントの繰返しプロセス
安全側面の開発は,ハザードの特定に基づくことが望ましい。規格開発の目標は,ハザードを特定し,
受容可能なリスクのレベルが達成されるように,リスクをコントロールするための手段を規定する規格を
作成することである。
危険状態に対するリスク分析,リスク評価及びリスクコントロールの繰返しプロセスは,受容可能なリ
スクを達成するために適したものである。規格作成者は,医療機器が開発から廃棄までのライフサイクル
全体を経ることに留意する必要がある。製品規格又はプロセス規格の作成者にとって重要な問題は,リス
クマネジメントの繰返しプロセスが,次の関係者によって確実に想定されるかどうかを判断することであ
る。
− 規格原案作成委員会:特定の及び既知のハザードについてリスク分析及びリスク評価を実施する(例
えば,規制上の適合性を実証するために使用される製品固有規格)。
− 規格の読者又はユーザー(例えば,製品の製造業者又は供給者):特定したハザード及び危険状態のリ
スク分析及びリスク評価を実施する。
規格作成者は,規格に含める適切な要求事項を決定する際に,これらの考慮事項を検討することが望ま
しい。
注記 規格が安全側面を導入している場合,規格は,特定の危険状態に関する安全要求事項だけを規定
することが可能である。このことは,その他の危険状態を特定し,それに関連するリスクをコン
トロールする製造業者の義務を除外するものではない。
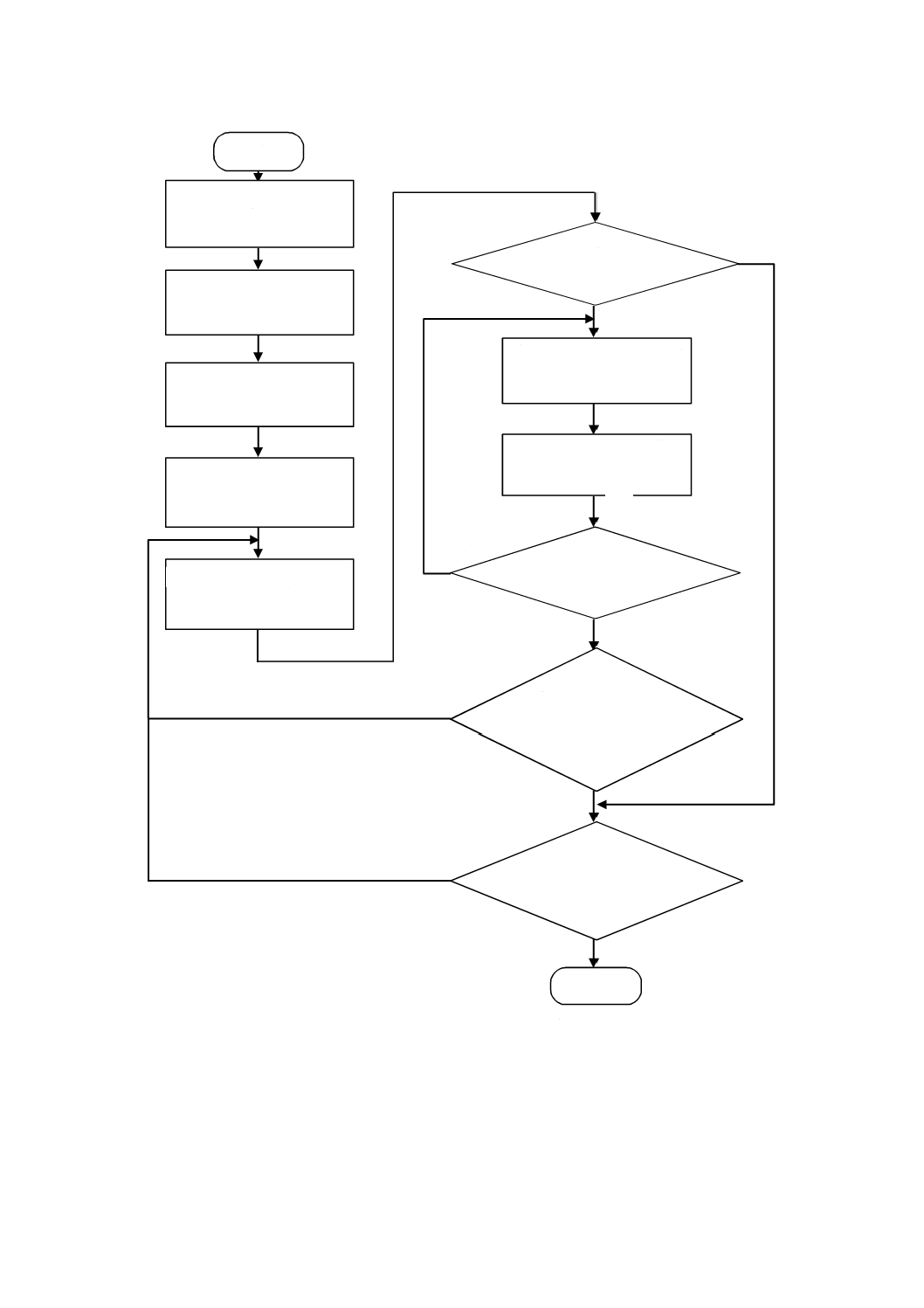
リスクを受容可能なレベルまで低減するために,次の手順に従うことが望ましい(図2)。
a) 製品の意図する使用及び合理的に予見可能な誤使用を特定する(7.3.3.1参照)。
b) 安全に影響を与えることがある製品又はプロセスの特質を明確化する(7.3.3.2参照)。
c) リスクの判断基準を確立し,受容可能なリスクレベルを決定する(7.2.3参照)。
d) 医療機器のライフサイクルにおいて(例えば,製品の使用,据付け,運転,保守,修理及び分解/廃
棄の状況において)発生する各ハザード(合理的に予見可能な危険状態を含む)を特定する(7.3.4参
照)。
e) 上記c)で定めたリスクの判断基準を使用して特定されたハザード及び危険状態に起因する,影響を受
16
T 0063:2020 (ISO/IEC Guide 63:2019)
けるユーザーグループに対するリスクを推定する(7.3.5参照)。
f)
上記c)で定めた受容可能なリスクレベルに基づき,リスクを評価する。この評価は,類似の医療機器
との比較によって行うことも可能である(7.3.6参照)。
g) リスクが受容可能でない場合は,リスク受容可能性の判断基準を満たすために,必要に応じて,各リ
スクを低減するリスクコントロール手段を特定する(7.3.7参照)。
h) 選択したリスクコントロール手段の有効性を検証する(7.3.8参照)。
i)
リスクコントロール手段を実施した後に残る残留リスクを推定し,評価する(7.3.9参照)。
j)
新しいハザード又は危険状態が生じたか,又は既存のリスクがリスクコントロール手段によって影響
されたかを決定する(7.3.10参照)。
k) 特定した全てのハザード(合理的に予見可能な危険状態を含む)が検討されたかを決定する(7.3.11参
照)。
箇条8に,安全側面を導入した医療機器規格が,リスクマネジメントの枠組みに適合するリスクマネジ
メントシステムの実施を,どのように促進できるかについての概要を示す。
17
T 0063:2020 (ISO/IEC Guide 63:2019)
図2−リスクマネジメント活動の繰返しプロセスの概要
7.3.3
意図する使用及び安全に影響することがある特質
7.3.3.1
意図する使用
対象とする製品又はプロセスの適用の決定には,次が含まれる。
a) 意図する医学的適応(例えば,検査,モニタ,治療,診断又は予防する,状態又は疾病)
開始
意図する使用を
特定する
[7.3.2 a)]
リスク低減は必要か
[7.3.2 f)]
安全に影響を与えることが
ある特質を明確化する
[7.3.2 b)]
適切なリスクコントロール
手段を特定する
[7.3.2 g)]
いいえ
はい
リスク判断基準を
確立する
[7.3.2 c)]
リスクコントロール手段の
有効性を検証する
[7.3.2 h)]
ハザード,危険状態,
事象を特定する
[7.3.2 d)]
いいえ
残留リスクは受容可能か
[7.3.2 i)]
各ハザード及び各危険状態に
対するリスクを推定する
[7.3.2 e)]
はい
新たな
ハザード又は危険状態が
発生するか又は既存のリスクに
影響を与えたか
[7.3.2 j)]
はい
いいえ
終了
はい
いいえ
全ての特定した
ハザードが検討されたか
[7.3.2 k)]
18
T 0063:2020 (ISO/IEC Guide 63:2019)
b) 意図する患者集団(例えば,年齢,体重,健康,容態)
c) 適用する又は対応する,意図する体の部分又は生体組織の種類
d) 意図するユーザープロファイル
e) 意図する環境条件及び使用方法
7.3.3.2
安全に影響することがある特質
製品又はプロセスの特質は,患者及びユーザーの安全に影響することがある。これらの特質は,規格の
必要性を検討するときに考慮することが望ましい。典型的な特質には,次が含まれる。
− 製造プロセス及びサービス手順(例えば,組立,校正)
− 設計プロセスのコントロール(例えば,ソフトウェア開発)
− 無菌性
− 生体適合性
− 発熱性
− 信頼性
− ユーザビリティ
− 機能性
− 感度及び特異度
− 環境影響
− 電気的安全
− 機械的強度
− 放射線安全
− 安定性
− 均質性
− データ完全性及びセキュリティ
ハザード及び危険状態を体系的に特定するためには,安全問題が発生し得る場合を定めるために,対象
とする製品又はプロセスを十分詳細に定義することが重要である。ただし,規格作成者は,一般的なハザ
ード及び危険状態だけしか特定できないことを理解することが望ましく,各製造業者が遭遇する全ての危
険状態を含めようとはしないことも望ましい。
7.3.4
ハザード及び危険状態の特定
7.3.4.1
ハザードの特定
意図する使用及び合理的に予見可能な誤使用に関連するハザードの特定には,通常,正常状態及び故障
状態の両方において製品に関連する既知の予見可能なハザードの特定を含む。
注記 故障状態に関連する事象は,医療機器のライフサイクルプロセスを含むことがある。ライフサイ
クルは,製品の設計,製造,据付け,サービス又は使用停止に関するプロセスを含むがこれに限
らない。
7.3.4.2
ハザード及び危険状態のタイプ
ハザードが特定されたら,次に危害の原因となり得る危険状態に至る一連の事象又は事象の組合せを検
19
T 0063:2020 (ISO/IEC Guide 63:2019)
討しなければならない。定義によれば,ハザードは,一連の事象又はその他の状況(通常使用を含む)が
ハザードに暴露される,すなわち,危険状態に至るまで危害をもたらすことはない(図1のP1)。危険状
態が存在しても,必ずしも,危害をもたらすとは限らない(図1のP2)。
7.3.4.3
医療機器関連のハザード及び危険状態
考えられる医療機器関連のハザードは,大部分が製品の性質に依存する。製品が故障しないことを保証
することは不可能であるため,規格は,医療機器のハザードを,患者の状態,健康及び安全並びにユーザ
ー及び他の人の健康及び安全に関わるものとして認識することが望ましい。ハザード及び危険状態を特定
する場合,規格作成者は,主として次に関連する危険状態に影響する要因を考慮することが望ましい。
a) 医療機器の設計,構造又は化学的安定性
b) 患者の状態及び患者環境
c) ユーザーによる医療機器の適用
d) 人が感染物質,危険な化学物質又は放射線などのハザードに暴露される可能性がある場合の,医療機
器の使用,サービス又は修理
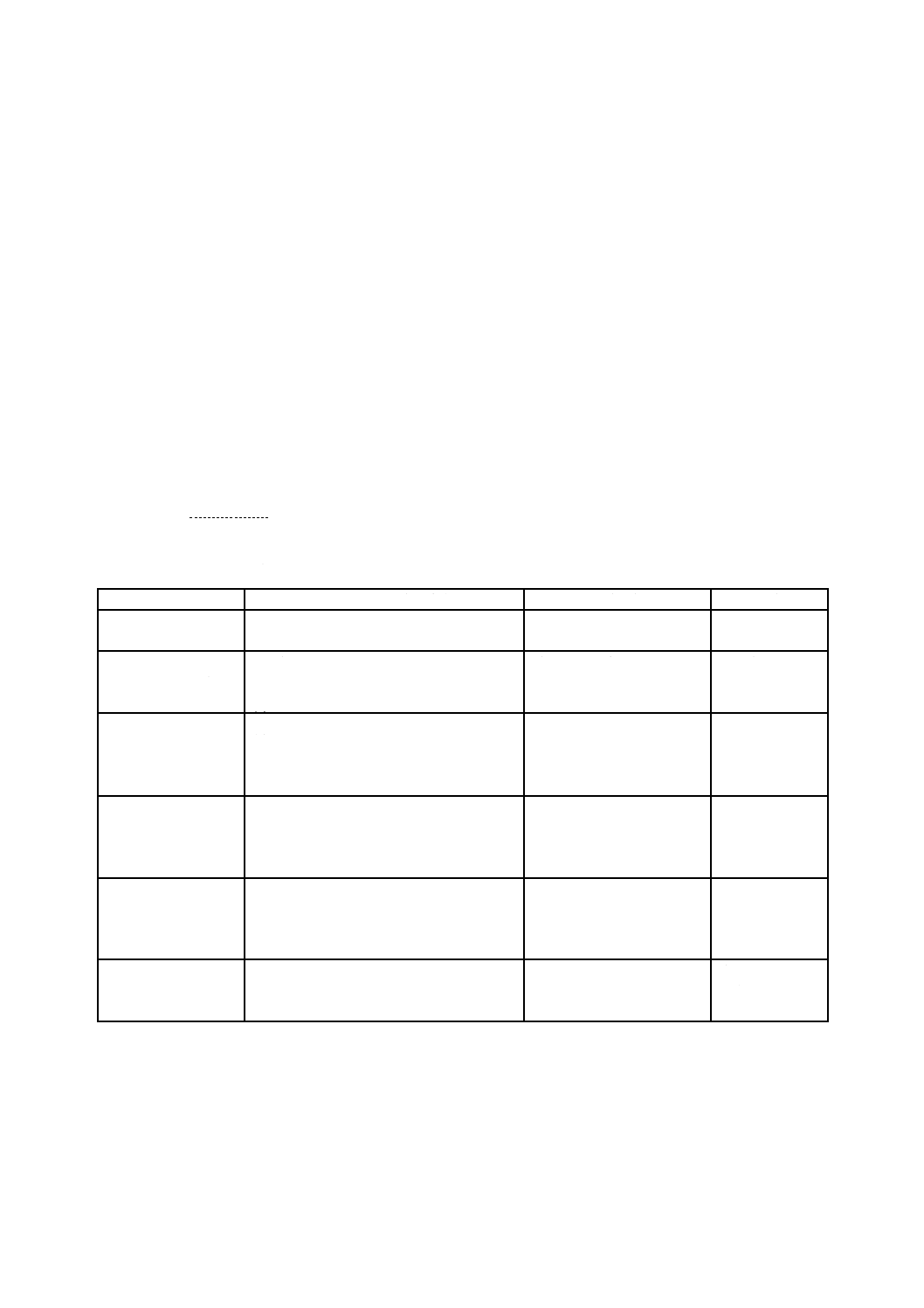
ハザード,予見可能な一連の事象,危険状態及び危害の間の関係を,幾つかの簡単な例について,表1
に示す。
表1−ハザード,予見可能な一連の事象,危険状態及び起こり得る危害の関係
ハザード
予見可能な一連の事象
危険状態
危害
電磁エネルギー
(高電圧)
(1) 電極ケーブルを意図せずに商用電源ソ
ケットに接続する。
商用電圧が電極上に生じ
る。
重篤な熱傷
心臓の細動
化学物質
(揮発性溶剤,塞栓)
(1) 製造中に使用した揮発性溶剤を完全に
除去しきれていない。
(2) 溶剤残留物が体温で気化する。
透析中に梗塞(血流中で気
泡が発生)が起こる。
ガス塞栓症
脳障害
生物学的
(微生物による汚染)
(1) 再使用麻酔チューブの浄化に関する指
示が不適切である。
(2) 麻酔時に汚染したチューブを使用す
る。
麻酔時に患者の気道内に細
菌が付着する。
細菌感染
機能性
(投与されない)
(1) 静電気に帯電した患者が注入ポンプに
触れる。
(2) 静電気放電(ESD)が原因となってポ
ンプ及びポンプアラームが故障する。
血糖値が上昇している患者
にインスリンが投与され
ず,警報も作動しない。
軽微な臓器障害
意識低下
機能性
(出力停止)
(1) 植込み型除細動器のバッテリーが寿命
に達する。
(2) 臨床的な経過観察受診間隔が不適切に
長い。
不整脈の発生時に除細動器
が作動しない。
死亡
測定
(不正確な情報)
(1) 測定エラー
(2) 測定エラーがユーザーによって検出さ
れない。
医師に不正確な情報が報告
され,誤診する,及び/又は
適切な治療が施されない。
疾病の進行
重傷
7.3.5
リスク推定
特定された危険状態について,関連するリスクを,入手できる情報又はデータを用いて推定する必要が
ある。一般に,危害の発生確率の定量的推定と定性的推定とで,いずれかが優れているということはない。
危害の発生確率の定量的推定は,有効な統計的データによって裏付けることができるものに限定される。
系統的な原因に関連する危害の発生確率は,推定することが困難なことがある(6.2.2参照)。
20
T 0063:2020 (ISO/IEC Guide 63:2019)
危害の発生確率を推定できない危険状態については,生じる可能性のある結果を考慮することが望まし
い。
この規格は,規格で取り扱う危険状態に関連するリスクを推定するために,規格作成者が特定のツール
又は方法を使用することは提言しない。
規格作成者が実施するリスク推定では,一連の信頼できる要求事項を得るために一般に認められた最新
の技術水準であるような入手可能なデータを考慮することが望ましい。情報源の参照及びそれらのデータ
を使用することの論理的根拠を,規格内に含めることが望ましい。
7.3.6
リスク評価
7.2.3で定めたリスク受容可能性の判断基準を使用し,規格作成者は,考慮対象のリスクのどれが規格の
要求事項を通じてコントロールされなければならないかを決定する。
7.3.7
リスクコントロールの特定
7.3.7.1
リスクコントロール手段の選択
選択したリスクコントロール手段は,製品の設計機能及びプロセスコントロールの両方を含むことが可
能である。分析した製品及び/又はプロセス,特定した結果としてのリスク,並びに,利用できるリスク
コントロール手段の有効性及び実現可能性に,その選択は依存する。規格の適用範囲には,これを反映す
ることが望ましい。選択したリスクコントロール手段は,安全の実用面に適切かつ一致するものである必
要がある(5.4参照)。
7.3.7.2
リスクコントロールに対する階層的アプローチ
リスクを受容可能なレベルまで低減することに適したリスクコントロール手段を決定する場合,次のリ
スクコントロールの選択肢を,記載の優先順位に従って適用しなければならない。
a) 設計による本質的安全
b) アラームを含む,医療機器自体の保護手段,又は製造若しくは保守に関連するプロセスのコントロー
ル
c) 安全に関する情報
規格作成者は,より低いレベルに移行する前に,この階層の各レベルにおける実施可能なリスクコント
ロールの選択肢を考慮しなければならない。
設計による本質的安全が,リスクコントロールプロセスにおける第一かつ最重要のステップである。経
験からは,十分に設計されたガード又は保護手段も故障する又は侵害され,安全に関する情報は必ずしも
守られてはいないことが知られているので,製品の特質に固有の設計ソリューションは引き続き有効な可
能性が高いためである。
設計による本質的安全の手段で,ハザードを排除することもリスクを十分にコントロールすることもで
きない場合は,ガード及び保護手段を考慮しなければならない。追加機器(例えば,非常停止機器)を含
む,補足的保護手段を実施することが必要なこともある。
最終ユーザーは,製造業者が提供する安全に関する情報又はトレーニングへの適合によって,リスクコ
ントロールを果たす役割をもつ。ただし,設計手段による本質的安全,ガード又は補足的保護手段を正し
く適用しないで,安全に関する情報又はトレーニングをその代替手段としてはならない。
21
T 0063:2020 (ISO/IEC Guide 63:2019)
7.3.7.3
適切なリスクコントロールとしての製品又はプロセス規格の要求事項
リスクをコントロールする必要がある場合,規格作成者は,製品又はプロセス規格における要求事項が
適切なリスクコントロールであるかを決定する。
製品規格は,可能な場合,規格作成者がリスクを受容可能なレベルまで低減する際に有効と考える検証
可能な技術的要求事項及び仕様を含んでいることが望ましい。
リスクコントロール手段は,危害の重大さを低減する若しくは危害の発生確率を低減する,又はその両
方を低減することが可能である。7.3.6で特定する受容できないリスクを取り扱うためにどのコントロール
手段を規格に含めることができるかを決定する場合,リスクをコントロールするために使用できる一般的
アプローチをまず考慮することが望ましい。7.3.7.2参照。
7.3.7.4
製品又はプロセス規格内のリスクコントロール手段としてのラベリング
7.3.7.4.1
情報の種類
規格は,製品に関与する人々(例えば,購入者,据付け業者,試験技術者,最終ユーザー及びサービス
要員)に提供する,意図する使用に必要な全ての情報について規定することが望ましい。
ラベリングの情報は,認知できる理解可能なもので,意図する使用環境における製品の正しい使用を支
援するものであることが望ましい。
規格作成者は,ラベリングによって提供される情報が,安全に関する情報又は残留リスクの開示である
かどうかを明確に特定することが望ましい。残留リスクの開示は,リスクコントロール手段ではない。
プロセス規格の場合,それらの規格は安全に関する情報の検証又はバリデーション活動を要求すること
があり得る。
製品規格の場合,それらの規格はどのような安全関連情報が必要かを,次のように明確に示すことが望
ましい。
− 製品自体及び/又は包装上に表示する。
− 販売時点で明確に視認できるようにする。
− 例えば,据付け,使用,保守及び廃棄について,説明資料を提供する。これにはトレーニング又は個
人用保護具の必要性に関する情報を含むことが望ましい。
関与する人が作業慣行に従うことでリスクが大幅に低減される場合は,適切な作業慣行についての情報
を記載することが望ましい。製品安全がかなりの程度で適切な作業慣行に依存する場合,また,これらの
慣行が自明のものでない場合,最低限,説明資料を参照するための表示を規定することが望ましい。
製品の使用に不可欠な安全に関する情報の価値を低下させる傾向があるため,不必要な情報は,避ける
ことが望ましい。
表示,記号及び安全標識(適切な記号又は安全標識が存在する場合)は,国際規格(例えば,ISO 3864
規格群,ISO 7000,ISO 7001,ISO 7010,ISO 15223-1及びIEC 60417)に従って規定することが望ましい。
7.3.7.4.2
説明書
規格は,提供される説明書及び情報が,製品又はシステムを作動するための必要な条件を対象とするこ
22
T 0063:2020 (ISO/IEC Guide 63:2019)
とを規定することが望ましい。
製品の場合,説明書は,組立,使用,洗浄,消毒,保守,解体,分解及び廃棄を適宜対象とする。
説明書の内容は,排除できない製品ハザードによって引き起こされる危害を防止するための手段を製品
のユーザーに示し,製品のユーザーが製品の使用に関する適切な決定をできるようにし,製品の誤使用を
避けるための指示を示すものであることが望ましい。
説明書には,製品が誤使用された場合(例えば,漂白剤を摂取した場合)の治療措置を指示することも
可能である。製品ハザードに関する説明書及び警告は,製品使用に関する指示と混同しないように個別に
書いて提示することが望ましい。
注記1 これに関連しては,ISO/IEC Guide 14,ISO/IEC Guide 37及びIEC Guide 109を参照。
注記2 取扱説明書の作成の原則は,IEC 82079-1に示されている。
7.3.7.4.3
警告
規格は,警告を次のように規定することが望ましい。
− 明白で,読みやすく,容易に消えず,かつ,理解しやすい。
− 特定の技術分野で別の言語で表すことが適している場合でない限り,製品又はシステムの使用を意図
されている国又は国々の公用語で表す。
− 簡潔かつ明確な分かりやすい文章とする。
警告には,一般的な又は特定の警告文を含めることが可能である。
警告は,使用が意図された全ての国の最終ユーザーに理解できるものであることが望ましい。
警告の内容は,製品ハザード,ハザードに起因する危害,及び警告を無視した場合に起こる結果につい
て記載することが望ましい。効果的な警告は,シグナル用語(“危険”,“警告”,“注意”という用語),安
全警告の図記号,製品のハザードに適した書体の大きさ及び色を使用し,注意を喚起する。適切な場合,
規格には,例えば,製品上,製品マニュアル内,安全データシート内といった警告の場所及び耐久性に関
する要求事項を含めることが望ましい。
さらに,試験方法を規定する規格には,手順及び/又は,例えば,試験所のスタッフにリスクをもたら
すことのある物質又は機器の使用を規定することが可能である。該当する場合,規格には,次のような警
告文を含めることが望ましい。
− 規格の最初に表す一般的警告文
例1 “注意−この規格に規定する試験の幾つかは,危険状態を招くことがあるプロセスの使用と
関わる。”
− 関連する規格の文章の前に適宜記載する特定の警告文
例2 “危険−非常に強い毒性をもつフルオロ酢酸ナトリウムの使用に起因する危険状態に注意す
る。”
7.3.7.5
包装
該当する場合,規格は,次のために,製品の包装に関する要求事項を規定することが望ましい。
− こん(梱)包された製品及び包装自体の適切な取扱い,輸送及び保管を確実なものにする。
23
T 0063:2020 (ISO/IEC Guide 63:2019)
− 製品の完全性を維持する。
− 傷害又は汚染に関連するようなハザードを除去し,又はそれらのリスクを最小限にする。
− 製品の適切な開こん(梱)を可能にする。
注記 これに関連しては,ISO/IEC Guide 41を参照。
7.3.8
有効性の検証
この規格に規定するリスクコントロール手段及び関連する試験方法は,規格を発行する前に,その有効
性を検証することが望ましい。この検証の一部は,科学文献から得ることができ,そうでなければ,規格
作成者が,試験所間調査の実施を決定することが可能である。
7.3.9
残留リスクの評価
一連のリスクコントロール手段を確立したら,実施した手段がリスクを受容可能にすることが期待でき
るかどうか決定することが望ましい。そうでない場合は,追加の又は代替のリスクコントロール手段を考
慮することが望ましい。この繰返し手順は,残留リスクが受容可能なレベルに低減されるまで継続するか,
又は規格のユーザーに残留リスクを通知して,ユーザーがリスクを更にコントロールするためにJIS T
14971を使用可能にすることが望ましい。
7.3.10 導入したリスクコントロール手段の影響
可能であれば,規格作成者は,導入したリスクコントロール手段の,この規格内の他のリスクコントロ
ール手段に対する影響,及びこれらの手段によって新しいリスクが発生するか又はそれらが他のリスクコ
ントロール手段の有効性に影響するかを検討することが望ましい。例えば,規格作成者は,リスクコント
ロール手段の医療機器のユーザビリティに対する影響を評価することが望ましい。
7.3.11 特定した全てのハザード及び危険状態に対する検討
規格作成者は,規格の作成中に特定されたハザード及び危険状態からの全てのリスクが検討されたこと
をレビューすることが望ましい。
7.4
規格のバリデーション
バリデーションは,規格を全ての利害関係者に回付して投票又はコメント募集を何度か繰り返し行う,
合意形成プロセスを通じて行う。
7.5
結論
上記のステップに従うことで,リスクに基づく体系的な製品規格又はプロセス規格の開発が促進される。
さらに,このアプローチは,JIS T 14971に適合するリスクマネジメントシステムの実施を促進する。
8
リスクマネジメントの枠組みにおける安全側面を導入した医療機器規格の適用の概要
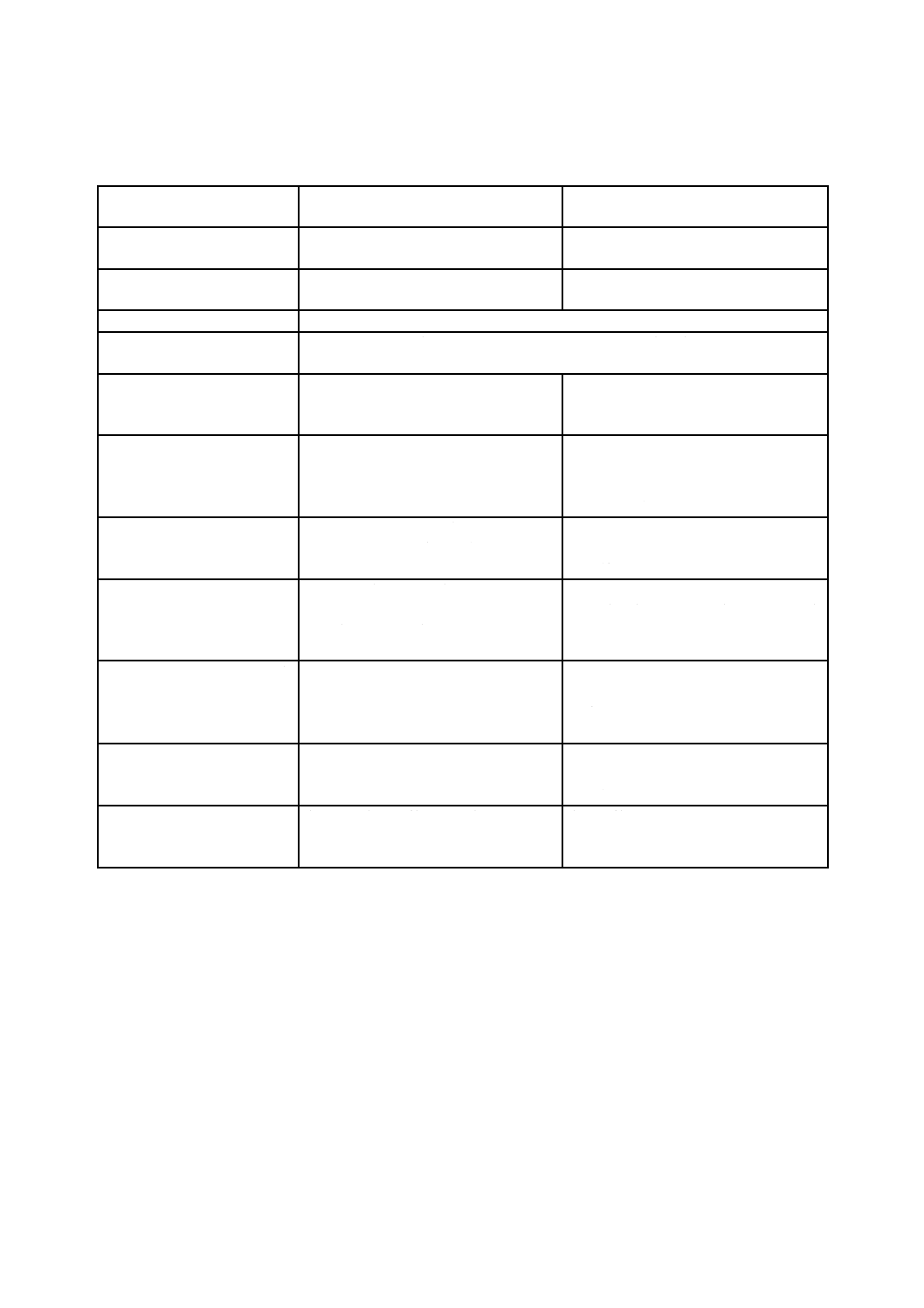
安全側面を導入したプロセス規格及び製品規格がどのようにリスクマネジメントの枠組みの実施を促進
するかの一般的指針については,表2を参照することが望ましい。プロセス規格又は製品安全規格が測定
可能なパラメーター及び受容可能な限度値を規定しない場合,製造業者が受容可能な限度値を開発するた
めの方法を規定する必要があり,又は適切なリスク受容可能性の判断基準を確立するためのリスクマネジ
メントの使用を実施することが望ましい。
24
T 0063:2020 (ISO/IEC Guide 63:2019)
表2−安全側面を導入した製品規格及びプロセス規格がリスクマネジメントプロセスの実施を
どのように促進できるかの指針
リスクマネジメントの
枠組みの要素
製品規格
プロセス規格
マネジメントの責任
部分的に適用してもよい。
マネジメントの追加的責任及びタスク
を定義してもよい。
リスクマネジメント計画
リスク受容可能性の判断基準を確立す
ることを手助けする。
リスクマネジメントの計画のための手
順を提供してもよい。
リスクマネジメントファイル
リスクマネジメントファイルに含める特定の内容を定義する。
意図する使用の特定
規格及び規格が対象とする安全に関連する特質が,取り扱う製品及びプロセスの意
図する使用及び合理的に予見可能な誤使用を定義してもよい。
ハザードの特定
医療機器のリスク分析で考慮するため
に,医療機器に関連する既知及び予見可
能なハザードを特定してもよい。
医療機器に関連する既知及び予見可能
なハザードを特定するための,正式な手
順を提供してもよい。
危険状態のリスクの推定
医療機器リスク分析で使用されること
がある危険状態が発生した場合の,危害
の重大さ又は危害の発生確率に関する
情報を提供してもよい。
危険状態が発生した場合に,危害の重大
さ又は危害の発生確率に関する情報を
体系的に収集するための,特定の情報又
は手順を提供してもよい。
リスク評価
規格内で定義された限度値は,通常,一
般に認められた最新の技術水準を反映
する。
リスクの受容可能性をどのように評価
し,決定するかについての方法又は手順
を提供してもよい。
リスクコントロール手段の選
択
リスク低減において有効であるとみな
される製造業者の医療機器設計におい
て実施できる,特定のリスクコントロー
ルの選択肢を提供してもよい。
製造業者が実施することができ,リスク
の低減に有効であると考えられる特定
のリスクコントロールの選択肢を提供
してもよい。
リスクコントロール手段の実
施
リスクコントロール手段の有効性を検
証する,特定の試験を提供してもよい。
製造業者が実施し,検証でき,リスクの
低減において有効であるとみなされる,
特定のリスクコントロール手段を提供
してもよい。
残留リスク評価
規格で定義された限度値は,通常,リス
クコントロールの一般に認められた最
新の技術水準を反映する。
リスクの受容可能性をどのように評価
し,決定するかについての方法又は手順
を提供してもよい。
製造及び製造後情報
製造及び製造後情報を収集するメカニ
ズムについて特定の要求事項を提供し
てもよい。
製造後情報の選択及び評価のための,及
びそれに続いて実施する是正措置のた
めの手順を提供してもよい。
25
T 0063:2020 (ISO/IEC Guide 63:2019)
附属書A
(参考)
製品安全規格及びプロセス安全規格
A.1 製品規格
一般に,安全な及び有効な医療機器に寄与する製品規格の場合は,統計的技法又はピアレビューなどの
科学的方法を使用して,試験所又は臨床試験に由来する科学的データに基づく必要がある。
製品規格は,一般に認められた最新の技術水準を表した,リスクを受容可能なレベルまでコントロール
する多様で健全な工学的手法を使用する。例には,次が含まれる。
− 人に対する物理的,化学的又は生物学的影響の安全上の限度値(X線量,電流値,表面温度,バイオ
バーデン,溶出物質量)
− 標準的な環境条件(温度及び湿度範囲,電磁界,特に制御された作動環境)
− 標準的なヒューマンインターフェイス(表示器,色,記号,アラームの考え方,文書化要求事項)
− 構造的詳細(電気絶縁,ケーブル接続)
− 適合性を実証するための標準的な試験(EMC試験,導電性テストフィンガー,生体適合性試験)
A.2 プロセス規格
プロセス規格が安全及び有効性に寄与できることは受け入れられているが,寄与の程度を定量化するこ
とは一般にはより困難である。ただし,それらは主に,コントロールされた方法で意図する結果を達成す
る(例えば,安全仕様を満たす)側面を重視し,一般に認められた最新の技術水準を表すことが可能であ
る。プロセス規格は,製品規格が一貫して実施されることを確実にすることにも寄与する。
プロセス規格は,設計,バリデーション,製造及びサービスをコントロールするプロセスを含めて,安
全な医療機器を作り上げるために不可欠なプロセスを標準化することによって,医療機器の安全に寄与可
能である(例えば,JIS T 14971,TR T 24971,JIS T 62366-1,JIS T 2304又はIEC TR 80002-1)。
特にリスクマネジメント目的で書かれたわけではないが,次は,リスクマネジメントを促進するプロセ
ス規格の例である。
− 製品製作プロセスの前提である最新の技術水準に基づく文書管理:これは一般に,品質マネジメント
システム規格(例えば,JIS Q 13485)によって取り扱われている。
− 生物学的試験を選択するための合意された枠組み(例えば,JIS T 0993)及び医療機器の滅菌プロセス
のための合意された方法(例えば,JIS T 0801又はJIS T 0806-1)。
26
T 0063:2020 (ISO/IEC Guide 63:2019)
附属書B
(参考)
リスク情報
規格作成者は,例えば,特定のハザードから生じる可能性のある危害,危険状態の結果としての危害の
発生確率,又は関連リスクに関係のある信頼性データに関するリスク情報を得るための情報源及び方法を
使用することが望ましい。このような情報は,外部情報源又は内部実験から得ることができる(データ収
集及び分析)。
リスク情報について考えられるデータ情報源は,次のとおりである。
− 医学的及び関連するデータベースを使用した査読付き文献の体系的レビュー
− 関連規格委員会からの技術論文
− 業界内の文書類などの文献
− 当該分野の専門家に知られた,その他の未発表情報源
− 発行された検査書に付帯する生データ
− 規制当局のデータベース
− 信頼性データベース
− 他の規格における参考文献
− 査読のない刊行物
− 検証されていないインターネット情報源(例えば,ソーシャルメディア)
情報源を選択する際は,慎重に行うことが望ましい。
27
T 0063:2020 (ISO/IEC Guide 63:2019)
参考文献
[1] ISO 3864 (all parts),Graphical symbols−Safety colours and safety signs
[2] ISO 7000,Graphical symbols for use on equipment−Registered symbols
[3] ISO 7001,Graphical symbols−Public information symbols
[4] ISO 7010,Graphical symbols−Safety colours and safety signs−Registered safety signs
[5] JIS Q 9000:2015 品質マネジメントシステム−基本及び用語
注記 対応国際規格では,ISO 9000:2015,Quality management systems−Fundamentals and vocabulary
を記載している。
[6] JIS T 0993(規格群) 医療機器の生物学的評価
注記 対応国際規格では,ISO 10993 (all parts),Biological evaluation of medical devicesを記載してい
る。
[7] JIS T 0801 ヘルスケア製品の滅菌−エチレンオキサイド−医療機器の滅菌プロセスの開発,バリデ
ーション及び日常管理の要求事項
注記 対応国際規格では,ISO 11135,Sterilization of health-care products−Ethylene oxide−Requirements
for the development, validation and routine control of a sterilization process for medical devicesを記
載している。
[8] JIS T 0806-1 ヘルスケア製品の滅菌−放射線−第1部:医療機器の滅菌プロセスの開発,バリデーシ
ョン及び日常管理の要求事項
注記 対応国際規格では,ISO 11137-1,Sterilization of health care products−Radiation−Part 1:
Requirements for development, validation and routine control of a sterilization process for medical
devicesを記載している。
[9] JIS Q 13485 医療機器−品質マネジメントシステム−規制目的のための要求事項
注記 対応国際規格では,ISO 13485,Medical devices−Quality management systems−Requirements for
regulatory purposesを記載している。
[10] JIS T 14971 医療機器−リスクマネジメントの医療機器への適用
注記 対応国際規格では,ISO 14971,Medical devices−Application of risk management to medical
devicesを記載している。
[11] ISO 15223-1,Medical devices−Symbols to be used with medical device labels, labelling and information to be
supplied−Part 1: General requirements
[12] TR T 24971 医療機器−JIS T 14971適用の指針
注記 対応国際規格では,ISO/TR 24971,Medical devices−Guidance on the application of ISO 14971
を記載している。
[13] IEC/TR 80002-1,Medical device software−Part 1: Guidance on the application of ISO 14971 to medical device
software
[14] JIS Z 8002 標準化及び関連活動−一般的な用語
注記 対応国際規格では,ISO/IEC Guide 2,Standardization and related activities−General vocabulary
を記載している。
[15] ISO/IEC Guide 14,Products and related services−Information for consumers
[16] ISO/IEC Guide 37,Instructions for use of products by consumers
28
T 0063:2020 (ISO/IEC Guide 63:2019)
[17] ISO/IEC Guide 41,Packaging−Recommendations for addressing consumer needs
[18] JIS Z 8051:2015 安全側面−規格への導入指針
注記 対応国際規格では,ISO/IEC Guide 51:2014,Safety aspects−Guidelines for their inclusion in
standardsを記載している。
[19] IEC Guide 109,Environmental aspects−Inclusion in electrotechnical product standards
[20] IEC 60417,Graphical symbols for use on equipment
[21] JIS T 2304 医療機器ソフトウェア−ソフトウェアライフサイクルプロセス
注記 対応国際規格では,IEC 62304,Medical device software−Software life cycle processesを記載し
ている。
[22] JIS T 62366-1:2019 医療機器−第1部:ユーザビリティエンジニアリングの医療機器への適用
注記 対応国際規格では,IEC 62366-1:2015,Medical devices−Part 1: Application of usability
engineering to medical devicesを記載している。
[23] IEC 80001-1:2010,Application of risk management for IT-networks incorporating medical devices−Part 1:
Roles, responsibilities and activities
[24] IEC 82079-1,Preparation of information for use (instructions for use) of products−Part 1: Principles and general
requirements
[25] GHTF/SG1/N055:2009,Definitions of the Terms “Manufacturer”, “Authorised Representative”, “Distributor”
and “Importer”
[26] GHTF/SG1/N071:2012,Definition of the Terms “Medical Device” and “In Vitro Diagnostic (IVD) Medical
Device”