K 5601-3-1 : 1999 (ISO 6503 : 1984)
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法に基づいて,日本工業標準調査会の審議を経て,通商産業大臣が改正した日
本工業規格である。
なお,この規格の制定後3か年を経た2002年4月をもって,この規格に対応するJIS K 5407(塗料成分
試験方法)は,廃止されこの規格に置き換わる予定であるので,なるべくこの規格によるとよい。
JIS K 5601は,次に示す部編成となっている。
JIS K 5601-1-1〜1-2 通則
JIS K 5601-2-1〜2-4 溶剤可溶物中の成分分析法
JIS K 5601-3-1 溶剤不溶物中の成分分析法
JIS K 5601-3は,塗料成分試験方法−溶剤不溶物中の成分分析法に関する試験方法として,次の各節に
よって構成する。
JIS K 5601-3-1 第3部−第1節:全鉛分(フレーム原子吸光分析法)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
K 5601-3-1 : 1999
(ISO 6503 : 1984)
塗料成分試験方法−
第3部:溶剤不溶物中の成分分析−
第1節:全鉛分(フレーム原子吸光分析法)
Testing methods for paint components−
Part 3 : Component analysis in solvent insoluble matter
Section 1 : Total leads (Flame atomic absorption spectrometric method)
序文 この規格は,1984年に第1版として発行されたISO 6503,Paints and varnishes−Determination of total
lead−Flame atomic absorption spectrometric methodを翻訳し,技術的内容及び規格票の様式を変更すること
なく作成した日本工業規格である。
なお,この規格で点線の下線を施してある箇所は,原国際規格にはない事項である。
1. 適用範囲 この規格は,塗料及び関連製品中の全鉛含有量を測定するための,フレーム原子吸光分析
法について規定する。この方法は,約0.01〜2% (m/m) の範囲の全鉛含有量をもつ製品に適用できる。
備考 この方法は,全鉛含有量が2% (m/m) 以上の製品にも適用できる。ただし,精度が7.2に示す
適切な数値を超えないときにだけ用いるべきである。
試料の処理については,二つの方法を示す。乾式灰化法(4.)は疑義が生じたときの審判分析法として使用
すべきである。
溶液中の鉛を測定する場合,ISO 3856-1に規定されているシチゾン吸光光度分析法を代替法として用い
てもよい。
2. 引用規格 次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成す
る。これらの引用規格のうちで,発行年を付記してあるものは,記載の年の版だけがこの規格の規定を構
成するものであって,その後の改正版・追補には適用しない。発行年(又は発効年)を付記していない引
用規格は,その最新版(追補を含む。)を適用する。
JIS K 0557 : 化学分析用の水
JIS K 5600-1-2 : 塗料一般試験方法−第1部:通則−第2節:試料採取方法
備考 ISO 1512 : 1991, Paints and varnishes−Sampling of products in liquid or paste formが,この規格
と一致している。
JIS K 5600-1-3 : 塗料一般試験方法−第1部:通則−第3節:試験用試料の検分及び調整
備考 ISO 1513 : 1992, Paints and varnishes−Examination and preparation of samples for testingが,この
規格と一致している。
2
K 5601-3-1 : 1999 (ISO 6503 : 1984)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ISO 385-1 Laboratory glassware−Burettes−Part 1 : General requirement
ISO 1042 Laboratory glassware−One-mark volumetric flasks
ISO 3856-1 Paints and varnishes−Determination of “soluble” metal content−Part 1 : Determination of lead
content−Flame atomic absorption spectrometric method and dithizone spectrophotrometric method
ISO 5725 Precision of test methods−Determination of repeatability by inter-labolatory tests
3. 原理 乾式灰化法(4.)又は湿式酸化法(5.)のいずれかによって試料を分解する。次いで,フレーム原子
吸光分析法によって鉛を分析する。
4. 乾式灰化法
4.1
原理 乾燥するまで試料を蒸発させ,あらゆる有機物質を除去するために475℃で灰化する。残留物
中のすべての鉛分を塩酸で抽出する。
4.2
試薬 分析は,分析試薬級の薬品及びJIS K 0557に規定するA2又はA3の水による。
4.2.1
無水炭酸ナトリウム
4.2.2
炭酸マグネシウム
4.2.3
硫黄
4.2.4
液状パラフィン
4.2.5
硫化ナトリウム 濃度10g/lの溶液。
4.2.6
塩酸 濃度約180g/lのもの。450mlの濃塩酸[36%(質量),密度約1.18g/ml]をほぼ同量の水に加
え,1 000mlまで希釈する。
4.2.7
塩酸 濃度約18g/lのもの。100mlの塩酸(4.2.6)を水に加えて1 000mlまで希釈する。
4.2.8
硝酸 濃度約315g/lのもの。硝酸[約65%(質量),密度約1.40g/ml]の1容を,2容の水に加え
る。
4.2.9
鉛標準保存溶液1l中に1g/lの鉛を含有する標準保存溶液 次のいずれかによって,調製する。
a) 正確に1gの鉛を含有する標準鉛溶液をアンプルに取り,その内容物を,あらかじめ少量の水と30ml
の硝酸(4.2.8)を入れた1 000mlの全量フラスコに移し,標線の位置まで水で希釈し,よく混合する。
b) あらかじめ105℃で2時間乾燥した硝酸鉛 [Pb(NO3)2] 1.598gを1mgのけたまではかり取る。これを1
000mlの全量フラスコ中で水に溶解する。30mlの硝酸(4.2.8)を加え,標線まで水で希釈し,よく混合
する。この標準保存溶液は1ml中に1mgの鉛を含有する。
4.2.10 鉛1l中に100mgの鉛を含有する標準溶液 この溶液は使用する日に調製する。10mlの標準保存溶
液(4.2.9)を100ml全量フラスコにピペットで取り,標線まで塩酸(4.2.7)で希釈し,よく混合する。
この標準溶液1mlは,100μgの鉛を含有する。
4.3
装置 通常の実験室の装置。
4.3.1
るつぼ シリカ製,望ましくは新品。
4.3.2
マッフル炉 475±25℃に維持できるもの。
4.3.3
ホットプレート 発熱量を制御できるもの。
4.4
試料採取 JIS K 5600-1-2の規定によって,試験を行う製品の代表サンプルを採取する。JIS K
5600-1-3の規定によって,試験用試料の検分及び調整を行う。
4.5
操作
3
K 5601-3-1 : 1999 (ISO 6503 : 1984)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4.5.1
予備試験 試験する製品の組成が未知であるならば,硝酸セルローズ又はアンチモンについて定性
試験を行う。これらの試験によって,硝酸セルローズ及びアンチモンが含まれないことが確認できなかっ
た場合,全操作を行う。
4.5.2
試料 この操作は2回繰り返して行う。試料を十分に混合し,直ちに,約5gを質量既知のるつぼ
(4.3.1)へ移す。試料は10mgのけたまではかる。
その製品が硝酸セルローズ(4.5.1)を含む場合は,るつぼ中の試料に,液状パラフィン(4.2.4)約2gを加え
て混ぜ合わせる。
4.5.3
灰化 ドラフト内で,試料を入れたるつぼをホットプレート(4.3.3)の上に載せる。徐々にホットプ
レートを昇温させ,揮発性溶剤をすべて除去する。
るつぼの内容物上に2gの炭酸マグネシウム(4.2.2)を散布し,るつぼを約350℃のマッフル炉(4.3.2)中,
10分間以上かけて徐々に入れ,さらに60分間以上かけて,475±25℃まで炉を昇温し,灰化が完了するま
でこの温度を保持する。酸化のための空気が適正に供給されていることを確かめる。るつぼ中の物質はい
かなる段階でも発火させてはいけない。
4.5.4
抽出
4.5.4.1
製品にアンチモンが含まれていない場合(4.5.1参照)は,次のように処理する。るつぼと灰分
(4.5.3参照)を冷却させる。るつぼと灰分を250mlのビーカーに移し,100mlの塩酸(4.2.6)を加える。ホ
ットプレート(4.3.3)を使って静かに15分間沸騰させる。さらに15分間加温を続けて分解する。まだ熱い
間に,き目の細かいろ紙(1)を通して,250mlビーカー中に傾斜ろ過する。
熱水でろ紙及び残留物を洗浄しビーカーに洗液を集める。ビーカーを冷却し,ろ液と洗液を250ml全量
フラスコに移す。標線まで水で希釈し,よく混合する。
注(1) 適当なろ紙の一例として,次のものがある。
Wattman No. 42及び44
Schleicher & Schull Nos. 589-3及び589-6
4.5.4.2
製品中にアンチモン(4.5.1参照)が含まれている場合は,次のように処理する。灰分(4.5.3参
照)を微細な粉末にし,それをもとのるつぼ内に戻し,炭酸ナトリウム(4.2.1)と硫黄(4.2.3)の等量合物,約
10gを加えて混合する。るつぼにふたをし,二酸化硫黄の臭いがなくなるまで,穏やかな炎で加熱する。
これには1〜2時間要するはずである。るつぼを冷却し,少量の熱水で,溶融物が完全に崩れるまで,内容
物を細かくほぐす。硫化ナトリウム溶液(4.2.5)を加えてろ過し,すべての残留物をろ紙へ移す。そして,
その残留物を硫化ナトリウム溶液で洗浄する。ろ液は捨てる。
ろ紙と残留物を250mlフラスコに移す。15mlの硝酸(4.2.8)を加え,ホットプレート(4.3.3)を用いて15分
間静かに沸騰する。100mlの塩酸(4.2.6参照)を加えて,30分間加温して分解する。まだ熱いうちに,き
目の細かいろ紙を通して,250mlのビーカーにろ過する。
次に熱水でろ紙と残留物を洗浄し,ビーカー内に洗液を集める。ビーカーを冷却し,ろ液と洗液を250ml
の定量フラスコに移す。水で標線まで希釈し,よく混合する。
4.5.5
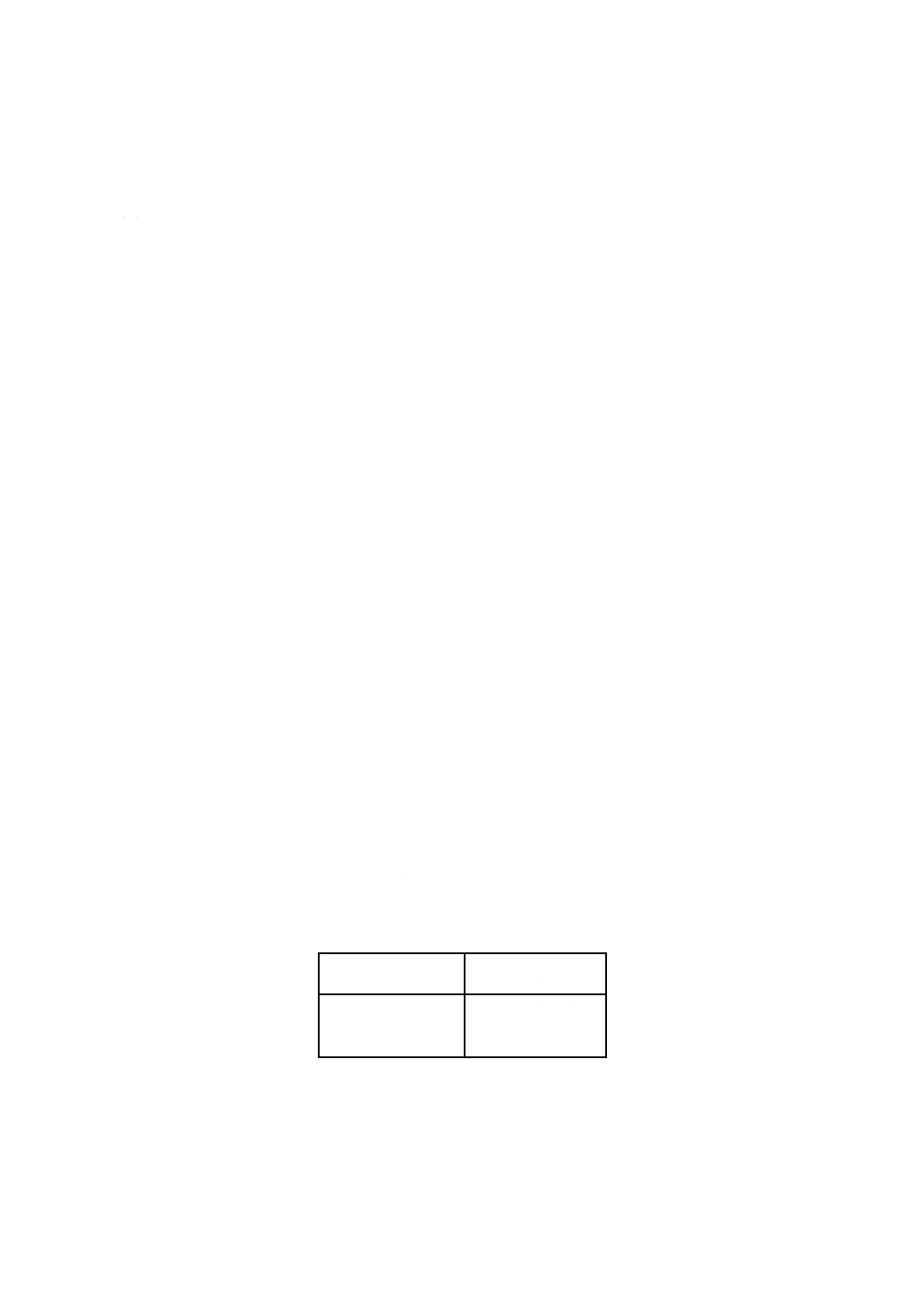
試験溶液の調製 表1に従って,試料中の予想される鉛含有量から,分取液量を決め,それぞれの
抽出液(4.5.4)から分取する。

4
K 5601-3-1 : 1999 (ISO 6503 : 1984)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表1
予想される鉛含有量
%(質量)
分取液量
ml
0.4%以下
25
0.4〜1
10
1〜2
5
備考 鉛量が2% (m/m) 以上であれば,適切
な分取液量を採取すべきである。
100mlの定量フラスコに分取液量(この場合は5mlの倍数)を採取する。それが5ml又は10mlであれば
10mlの塩酸(4.2.6)を加える。水で標線まで希釈してよく混合する。
4.5.6
試料空試験 試料を除いて,4.5.3,4.5.4.1,又は4.5.4.2及び4.5.5の操作を繰り返す。
5. 湿式酸化法
5.1
原理 すべての有機物質を除去するため,ビーカー中で硫酸と過酸化水素との混合物によって(方
法A),又はケルダールフラスコ中で硫酸と硝酸との混合物によって(方法B),試料を湿式酸化する。過
剰な硫酸を取り除くため加熱し,次に残留物中のすべての鉛分(硫酸鉛の形で)をEDTAとアンモニア溶
液で抽出する。
備考 試料中のアンチモン又は硝酸セルローズの存在は,この方法では妨害にならない。
5.2
試薬 分析には,分析試薬級の薬品及びJIS K 0557に規定するA2又はA3の水による。
5.2.1
硫酸 硫酸約96%(質量),密度約1.84g/mlのもの。
5.2.2
過酸化水素 約30%(質量),又は適当な安全対策をとった場合は,約50%(質量)溶液のもの。
5.2.3
硝酸 濃度約65%(質量),密度約1.40g/mlのもの。
5.2.4
硝酸 濃度約315g/ml硝酸(5.2.3参照)1容を,2容の水に加える。
5.2.5
アンモニア溶液 濃度約85gNH3/l溶液。濃アンモニア溶液[25%(重量)]380mlを水で1 000ml
に希釈する。
5.2.6
EDTA(エチレンジアミン四酢酸二ナトリウム) 37g/l溶液のもの。
5.2.7
鉛 1l中に1gの鉛を含有する標準保存溶液
a) 1gの鉛を含有する標準鉛溶液のアンプルの内容物を,あらかじめ少量の水及び30mlの硝酸(5.2.4)を入
れた1 000mlの全量フラスコに移し,標線の位置まで水で希釈し,よく混合する。
b) あらかじめ105℃で2時間乾燥した硝酸鉛 [Pb(NO3)2] 1.598gを1mgのけたまではかり取る。これを1
000mlの定量フラスコ中で水に溶解する。30mlの硝酸(5.2.4参照)を加え,水で標線まで希釈し,よ
く混合する。
この標準保存溶液1mlは,1mgのPbを含有する。
5.2.8
鉛1l中に100mgのPbを含有する標準溶液 この溶液は使用する日に調製する。10mlの標準保存
溶液(5.2.7)をピペットで取り,100ml定量フラスコに入れ,10mlの硝酸(5.2.4)を加え,標線まで水で希釈し,
よく混合する。この標準溶液1mlは,100μgの鉛を含有する。
5.3
装置 通常の実験室の装置
5.3.1
ホットプレート 発熱量を制御できるもの。
5.4
試料採取 JIS K 5600-1-2の規定によって試験を行う製品の代表試料を採取する。JIS K 5600-1-3の
規定によって,試験用試料の検分及び調整を行う。
5.5
操作
5
K 5601-3-1 : 1999 (ISO 6503 : 1984)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5.5.1
試料 この操作は,2回繰り返す。試料を完全に混合し,直ちに約0.5gを400mlのビーカー(方法
A),又は250mlのケルダールフラスコ(方法B)に移す。試料は0.1mgのけたまではかる。
5.5.2
分解
5.5.2.1
方法A ドラフト内で,試料を入れたビーカーをホットプレート(5.3.1参照)上に載せる。すべ
ての揮発性溶剤を除去するために静かに加熱する。約5mlの硫酸(5.2.1参照)を加え,時計皿でビーカー
を覆い,約15分間,より高い温度で加熱する。有機物質を分解・炭化する。白煙が発生するまで加熱を継
続する。ビーカーをホットプレートから取り出し,約10分間冷却する。5mlのピペットを用いて,過酸化
水素溶液(5.2.2参照)を5mlずつ4回ゆっくりと加える。毎回の添加は,反応が静まるのを待って行う(警
告参照)。
警告 飛散の危険があるため,過酸化水素溶液の(4回の)添加の間は,ビーカーに,カバー(時計皿)
をしたままにしておく。
再び10分間加熱し,5分間冷却する。それから過酸化水素溶液5mlずつをさらに2回加える。5分間加
熱し,5分間冷却する。最後に,さらに過酸化水素溶液5mlを加える。残存している過酸化水素を分解す
るために,再び加熱する。時計皿を取り除き,注意深く水で底面を洗い落とす。洗液はビーカーに採取す
る。著しい白煙が発生するまでビーカーを加熱する。次いでその溶液が,ほとんど乾燥するまで蒸発させ
る。ホットプレートからビーカーを下ろし,冷却する。
5.5.2.2
方法B すべての揮発性溶剤を取り除くために,ケルダールフラスコとその内容物をブンゼンバ
ーナーで緩やかに加熱する。約5mlの硫酸(5.2.1)を加え,有機物質を分解,炭化させるために約10分間加
熱し,約10分間冷却する。5mlのピペットを用いて硝酸を(5.2.3)5mlずつを4回,ゆっくりと加える。毎
回の添加は反応が静まるのを待って行う。
再び約10分間加熱し,5分間冷却する。次いで硝酸を5mlずつをさらに2回加える。5分間加熱し,5
分間冷却する。最後に,さらに硝酸5mlを加える。すべての硝酸を分解するために,著しい白煙が発生す
るまで,再び加熱し,溶液がほとんど乾燥するまで蒸発させ,冷却する。最後の段階で炭化が起こってい
れば,注意しながら硝酸をさらに加え,加熱,白煙発生,冷却の操作を繰り返す。
5.5.3
抽出 ビーカー(方法A)又はケルダールフラスコ(方法B)に50mlのEDTA溶液(5.2.6),10ml
のアンモニア溶液(5.2.5),及び50mlの水を加える。静かに約15分間煮沸し,冷却する。もし必要ならば,
中程度の目の粗さのろ紙を使って250mlの全量フラスコ中に傾斜ろ過し,ろ紙及び残留物を水洗した洗液
を加え,標線まで水で希釈し,よく混合する。
5.5.4
表2に従って試料中の予想される鉛含有量から分取液を決め,それぞれの抽出液(5.5.3)から分取す
る。分取量は,表2に従って試料中の予想される鉛含有量から求める。
表2
予想される鉛含有量
%(重量)
分取量
ml
0.5以下
100(無希釈)
0.5〜1
75
1〜2
50
備考 鉛含有量が2% (m/m) 以上であれば,
適切な分取液量を採取すべきである。
100mlの定量フラスコに,その分取液を入れ,水で標線まで希釈し,よく混合する。
5.5.5
試薬空試験 試料を除いて,5.5.2.1(A法)又は5.5.2.2(B法),5.5.3及び5.5.4の操作を繰り返す。
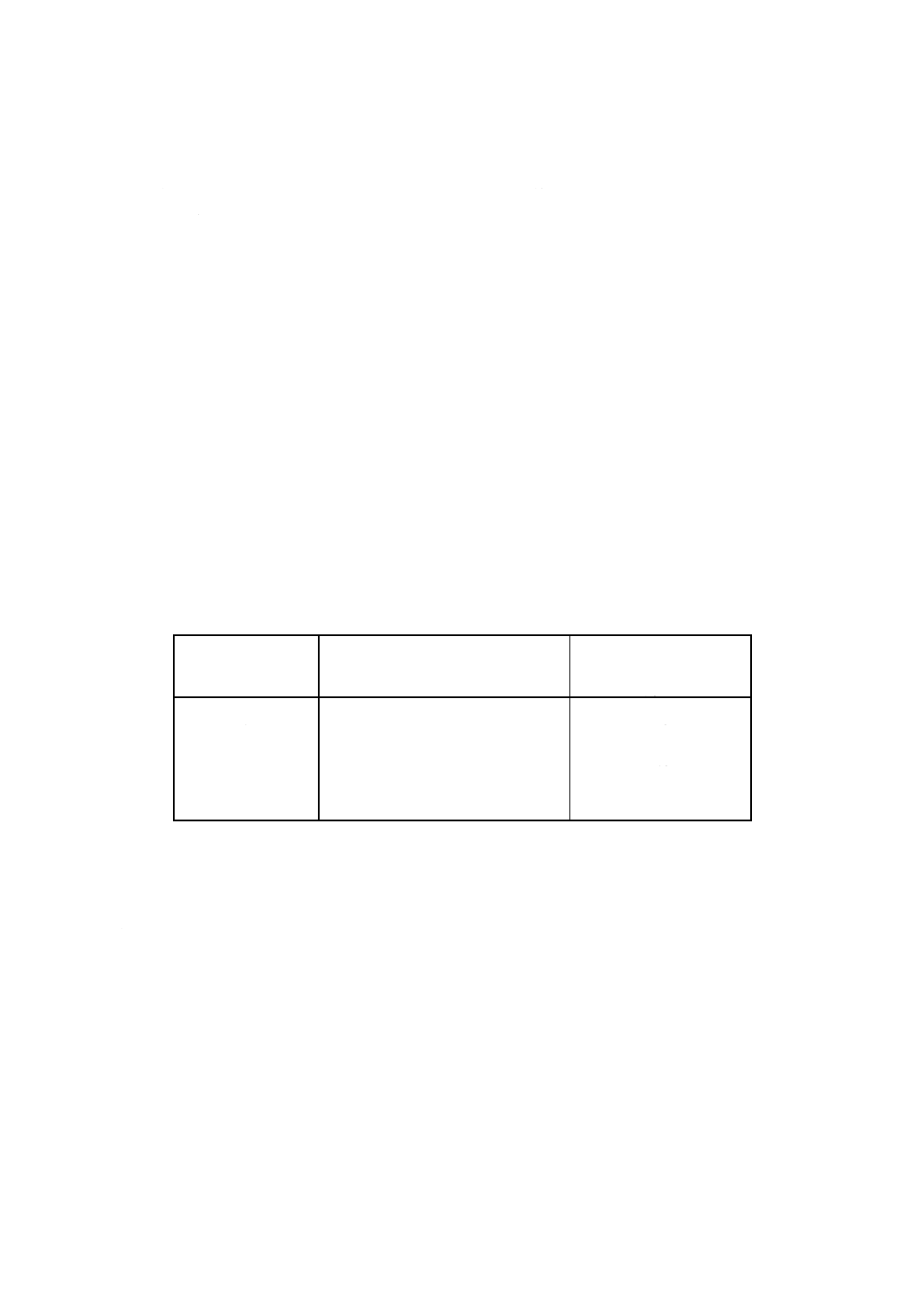
6
K 5601-3-1 : 1999 (ISO 6503 : 1984)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6. 定量
6.1
原理 試験溶液をアセチレン/空気フレーム中に噴霧する。鉛用中空陰極ランプ又は鉛用放電ラン
プから放射される波長283.3nmの領域に選定されたスペクトル線の吸光度を測定する。
6.2
試薬及び材料 分析には,分析試薬級の試薬及び蒸留水,又は同等の純度の水を用いる。
6.2.1
アセチレン 鋼製円筒容器に入った市販のもの。
6.2.2
圧縮空気
6.3
装置 通常の実験室の装置
6.3.1
フレーム原子吸光分析装置 波長283.3nmでの測定に適し,アセチレンと空気の燃焼バーナーを備
えたもの。
6.3.2
鉛用中空陰極ランプ又は鉛用放電ランプ
6.3.3
ビュレット 容量50ml,ISO 385-1の要求事項に適合するもの。
6.3.4
全量フラスコ 容量100ml,ISO 1042の要求事項に適合するもの。
6.4
操作
6.4.1
検量線の作成 次に示す検量線を作成するか,少なくとも同等の正確さ及び精度をもつ機器を用い
る。
6.4.1.1
標準検量溶液の調製 これらの溶液は使用する日に調製する。6個の100mlの全量フラスコ(6.3.4)
に,それぞれビュレット(6.3.3)を用いて,表3に示す量の標準鉛溶液(4.2.10又は5.2.8)をはかり取る。
10mlの塩酸(4.2.6)を加え,標線まで水で希釈し,よく混合する。
表3
標準検量溶液の番号
標準鉛溶液
標準検量溶液中の鉛の濃度
(4.2.10又は適切ならば5.2.8の容量)
ml
μg/ml
0*
0
0
1
2
2
2
5
5
3
10
10
4
20
20
5
30
30
*
ブランク検量溶液
6.4.1.2 分光測定 鉛用光源ランプ(6.3.2)を分光器(6.3.1)に取り付ける。鉛定量のための条件を最適化する。
装置を装置製造業者の手順書に従って調整し,最大の吸光度が得られるように,モノクロメーターを
283.3nmの領域に調節する。噴霧器及びバーナーの性質に従って,アセチレン(6.2.1)と空気(6.2.2)の流量を
調節し,点火してフレームを形成する。もし調整できるならば,標準検量溶液のNo.5(表3参照)におい
て吸光度がほぼフルスケールの値を示すように,スケールの拡大率を設定する。
それぞれの標準検量溶液(6.4.1.1)を,濃度の低い方から順にフレームの中に噴霧,測定し,装置が安定性
を保っていることを確認するために,標準検量溶液No.4を用いて測定を繰り返す。個々の測定の間には,
噴霧の状態を一定に保つように注意しながら,バーナーを通して水を噴霧する。
6.4.1.3
検量線 それぞれの標準検量溶液1ml中に含まれる鉛の量をマイクログラム単位でグラフの横座
標にとり,対応する吸光度からブランク検量溶液の吸光度を差し引いた値を縦座標にプロットする。
6.4.2
定量測定
7
K 5601-3-1 : 1999 (ISO 6503 : 1984)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6.4.2.1
6.4.1.2の手順で,分光器(6.3.1)を調整した後,まずブランク試験溶液(4.5.6又は5.5.5)の吸光度
を測定する。次に個々の試験溶液(4.5.5又は5.5.4)の吸光度を3回ずつ測定し,再びブランク試験溶液の
吸光度を測定する。最後に,装置の応答状態が変わっていないことを確認するため,再度,標準検量溶液
No.4(表3参照)の吸光度を測定する。もし試験溶液の吸光度が最も高い鉛濃度をもつ標準検量溶液の吸
光度よりも高い場合は,試験溶液を適宜(希釈係数をFとする),既知量の水で希釈する。
6.4.2.2
一つの試験溶液について,(3回の)読取り値が2%以上異なるか,平均値より1%以上異なる場
合は,6.4.2.1の操作を繰り返す。
6.4.2.3
試薬の空試験値を補正し,吸光度から検量線を用いて個々の試験溶液中の鉛濃度cを求める。直
読式の測定装置によって自記された鉛濃度を記録する。
7. 結果の表し方
7.1
計算 塗料の全鉛含有量は,次の式によって算出する。
V
m
F
c
a
×
×
×
5.2
=
ここに,
a: 塗料中の全鉛含有量(質量%)
c: 検量グラフから得られた試験溶液中の鉛濃度 (μg/ml)
F: 6.4.2.1に示す希釈係数
m: 試料(4.5.2又は5.5.1)の質量 (g)
V: 4.5.5又は5.5.4において分取した抽出液の容量 (ml)
2個の測定値の平均を求める。
7.2
精度
備考 次の値は,得られた測定値を用い,ISO 5725に規定する計算法に基づいて求めたものである。
これらの値は,操作について経験を積んだ場合,修正されることがある。
7.2.1
繰返し精度 (r) 同一の試料について,短時間内に同一の操作者が同一の実験室で同一の装置を用
いて,標準試験方法で行った2回の試験結果の差の絶対値が,95%の確率で存在すると期待される範囲の
限界値は次のとおりである。
5% 4.5.4.1に記載した,乾式灰化法における,平均値に対する相対%。
10% 4.5.4.2に記載した,アンチモンを含有する製品を対象にした,乾式灰化後アセチレン抽出方法
における,平均値に対する相対%。
5% 4.5.2.1に記載した,湿式酸化法における,平均値に対する相対%(7.2.2の備考2.参照。)。
7.2.2
再現精度 (R) 同一の試料について,異なった試験室で,異なった操作者が,標準化試験方法で
行った2回の試験結果間の差の絶対値が,95%の確率で存在すると期待される範囲の限界値は次のとおり
である。
15% 4.5.4.1に記載した,乾式灰化方法における,平均値に対する相対%。
30% 4.5.4.2に記載した,アンチモンを含有する製品を対象にした,乾式灰化後アセチレン抽出方法
における,平均値に対する相対%。
15% 4.5.2.1に記載した,湿式酸化法における,平均値に対する相対%(備考参照)。
備考1. 5.5.2.1(A法)で述べた湿式酸化法について提供された精度データは,単一の塗料試料に関
するものである。
2. 5.5.2.2(B法)で述べた湿式酸化法については,データが得られていない。
8
K 5601-3-1 : 1999 (ISO 6503 : 1984)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8. 試験報告 試験報告には,少なくとも次の事項を含んでいなければならない。
a) 試験した製品の種別及びその明細
b) この規格の適用
c) 製品中の鉛を抽出するために用いた前処理法(乾式灰化又は湿式酸化),湿式酸化法を用いた場合,利
用した分解法(A法又はB法)を記録
d) 4.5.2に従って実施した,いずれかの予備処理法
e) 試験結果
f)
受渡当事者間の協定又は別の方法によって,規定した試験手順を変更した場合,その内容
g) 試験年月日
塗料分野の国際整合化調査研究委員会 構成表
氏名
所属
(委員長)
増 子 昇
千葉工業大学
(委員)
西 出 徹 雄
通商産業省基礎産業局
大 嶋 清 治
工業技術院標準部
鴨志田 直 史
工業技術院標準部
橋 本 繁 晴
財団法人日本規格協会
本 橋 健 司
建設省建築研究所
坪 田 実
職業能力開発大学校
武 井 昇
職業能力開発大学校
鈴 木 雅 洋
東京都立産業技術研究所
吉 田 豊 彦
社団法人色材協会
高 橋 孝 治
社団法人日本塗装工業会
青 木 茂
サンコウ電子研究所
福 島 稔
社団法人日本鋼橋塗装専門会
近 藤 照 夫
清水建設株式会社
(主査)
岩 井 弘
財団法人日本検査協会
堀 江 建 治
関西ペイント株式会社
山 田 俊 幸
神東塗料株式会社
中 東 昭 憲
神東塗料株式会社
住 田 光 正
大日本塗料株式会社
上 寺 孝 明
中国塗料株式会社
松 井 繁 武
株式会社トウペ
更 谷 浩
日本特殊塗料株式会社
曽 我 元 昭
日本ペイント株式会社
大 澤 晃
日本油脂株式会社
高 橋 真
ロックペイント株式会社
長 尾 進
専門技術者
鈴 木 幹 夫
専門技術者
松 平 忠 志
松平技術士事務所
伊 藤 義 人
専門技術者
小 島 務
財団法人日本検査協会
常 田 和 義
大日本塗料株式会社
筒 井 晃 一
日本ペイント株式会社
(事務局)
内 田 幹 雄
社団法人日本塗料工業会
山 崎 不二雄
社団法人日本塗料工業会
文責 松井繁武