H 1283 : 1999
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法に基づいて,日本工業標準調査会の審議を経て,通商産業大臣が制定した日
本工業規格である。
今回の新規制定に当たっては,日本工業規格と国際規格との対比,国際規格に一致した日本工業規格の
作成及び日本工業規格を基礎にした国際規格の原案の提案を容易にするため,ISO 6351 : 1985, Nickel−
Determination of silver, bismuth, cadmium, cobalt, copper, iron, manganese, lead and zinc content−Flame atomic
absorption spectrometric method, ISO 7530-1 : 1990, Nickel alloys−Flame atomic absorption spectrometric
analysis−Part 1 : General requirements and sample dissolution, ISO 7530-2 : 1990, Nickel alloys−Flame atomic
absorption spectrometric analysis−Part 2 : Determination of cobalt content 及びISO 9389 : 1989, Nickel alloys−
Determination of cobalt content−Potentiometric titration method with potassium hexacyanoferrate(III)を基に翻
訳し,技術的内容を変更することなく作成した。
この規格の一部が,技術的性質をもつ特許権,出願公開後の特許出願,実用新案権,又は出願公開後の
実用新案登録出願に抵触する可能性があることに注意を喚起する。通商産業大臣及び日本工業標準調査会
は,このような技術的性質をもつ特許権,出願公開後の特許出願,実用新案権,又は出願公開後の実用新
案登録出願にかかわる確認について,責任はもたない。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
H 1283 : 1999
ニッケル及びニッケル合金中の
コバルト定量方法
Methods for determination of cobalt nickel and nickel alloys
序文 この規格は,対応国際規格であるISO 6351 : 1985, Nickel−Determination of silver, bismuth, cadmium,
cobalt, copper, iron, manganese, lead and zinc content−Flame atomic absorption spectrometric method, ISO
7530-1 : 1990, Nickel alloys−Flame atomic absorption spectrometric analysis−Part 1 : General requirements and
sample dissolution, ISO 7530-2 : 1990, Nickel alloys−Flame atomic absorption spectrometric analysis−Part 2 :
Determination of cobalt content及びISO 9389 : 1989 , Nickel alloys−Determination of cobalt content−
Potentiometric titration method with potassium hexacyanoferrate(III)の対応する部分(ISO 6351については,
コバルトの定量に関する部分,ISO 7530-2及びISO 9389については全体)と技術的内容を変更すること
なく作成した。
なお,対応国際規格がない定量方法(銅分離ニトロソR塩吸光光度法)を日本工業規格として追加規定し
ている。
1. 適用範囲 この規格は,ニッケル及びニッケル合金中のコバルトの定量について規定する。
備考 この規格の対応国際規格を,次に示す。
ISO 6351 : 1985 Nickel−Determination of silver, bismuth, cadmium, cobalt, copper, iron, manganese,
lead and zinc content−Flame atomic absorption spectrometric method
ISO 7530-1 : 1990 Nickel alloys−Flame atomic absorption spectrometric analysis−Part 1 : General
requirements and sample dissolution
ISO 7530-2 : 1990 Nickel alloys−Flame atomic absorption spectrometric analysis−Part 2 :
Determination of cobalt content
ISO 9389 : 1989 Nickel alloys−Determination of cobalt content−Potentiometric titration method
with potassium hexacyanoferrate(III)
2. 引用規格 次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成す
る。この引用規格は,その最新版を適用する。
JIS H 1270 ニッケル及びニッケル合金の分析方法通則
JIS H 1272 ニッケル及びニッケル合金中の銅定量方法
JIS K 8005 容量分析用標準物質
2
H 1283 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3. 一般事項 分析方法に共通な一般事項は,JIS H 1270の規定による。
4. 定量方法の区分 コバルトの定量方法は,次のいずれかによることとし,各定量方法の適用試料は,
表1による。
a) 銅分離ニトロソR塩吸光光度法 この方法は,コバルト含有率0.02% (m/m) 以上1.0% (m/m) 以下の
試料に適用する。
b) ヘキサシアノ鉄(III)酸カリウム滴定法 この方法は,コバルト含有率2% (m/m) 以上25% (m/m) 以
下の試料に適用する。
c) 原子吸光法(A法) この方法は,コバルト含有率0.01% (m/m) 以上2.0% (m/m) 以下の試料に適用
する。
d) 原子吸光法(B法) この方法は,コバルト含有率0.01% (m/m) 以上4.0% (m/m) 以下の試料に適用
する。
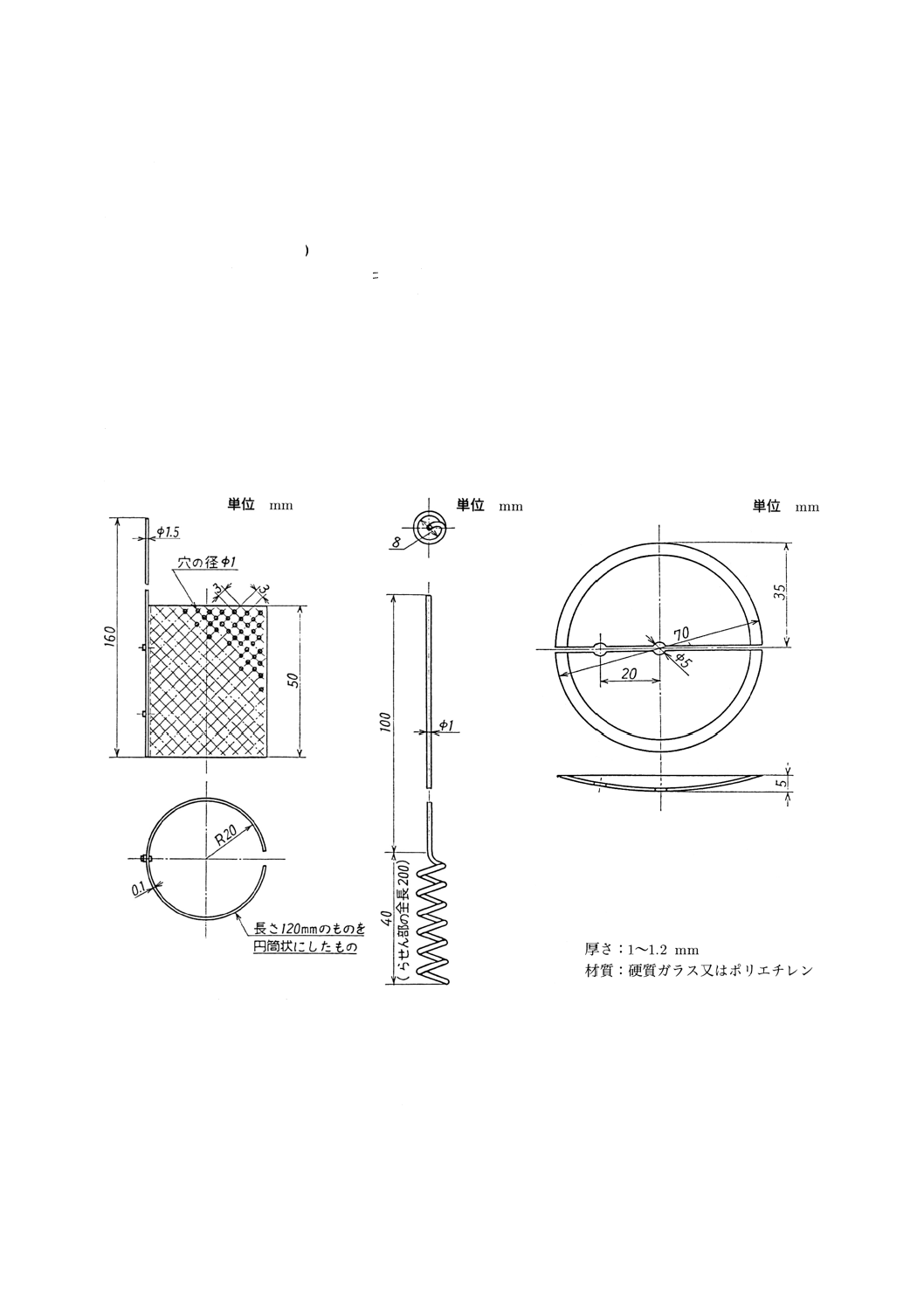
表1 定量方法及び適用試料
試料
銅分離ニトロソ
R塩吸光
光度法
ヘキサシアノ鉄
(III)
酸カリウム
滴定法
原子吸光法
(A法)
原子吸光法
(B法)
合金番号
合金記号
NW2200
Ni99.0
−
−
○
○
NW2201
Ni99.0-LC
−
−
○
○
NATB
−
−
−
○
NW4400
NiCu30
○
−
−
○
NW4402
NiCu30-LC
○
−
−
○
NW5500
NiCu30A13Ti
○
−
−
○
NW0001
NiMo30Fe5
−
○
−
○
NW0665
NiMo28
−
−
−
○
NW0276
NiMo16Cr15Fe6W4
−
○
−
○
NW6455
NiCr16Mo16Ti
−
−
−
○
NW6022
NiCr21Mo13Fe4W3
−
○
−
○
NW6007
NiCr22Fe20Mo6Cu2Nb
−
○
−
○
NW6985
NiCr22Fe20Mo7Cu
−
○
−
○
NW6002
NiCr21Fe18Mo9
−
○
−
−
ニッケル鋳物
−
−
○
○
ニッケル−銅合金鋳物
○
−
−
○
ニッケル−モリブデン合金鋳物
−
−
−
○
ニッケル−モリブデン−クロム合金鋳
物
−
−
−
○
ニッケル−クロム−鉄合金鋳物
−
−
−
○
5. 銅分離ニトロソR塩吸光光度法
5.1
要旨 試料を硝酸と硫酸との混酸で分解し,銅を電解して分離した後,溶液を蒸発乾固する。水と
硫酸を加えて可溶性塩類を溶解し,酢酸ナトリウムとニトロソR塩を加え,煮沸してコバルトを呈色させ,
硝酸を加えて煮沸した後,光度計を用いて,その吸光度を測定する。
5.2
試薬 試薬は,次による。
a) 硝酸 (1+1)
3
H 1283 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) 硫酸 (1+3)
c) 混酸 水400mlに硫酸100mlを少量ずつかき混ぜながら加え,常温まで冷却した後,硝酸100mlを加
える。
d) 酢酸ナトリウム溶液 酢酸ナトリウム三水和物30gを水に溶解し,水で液量を100mlとする。
e) ニトロソR塩溶液 (7.5g/l) この溶液は,使用の都度調製する。
f)
標準コバルト溶液 (10μgCo/ml) コバルト [99.9% (m/m)] 0.100gをはかり取ってビーカー (100ml)
に移し入れ,硝酸 (1+1) 10mlを加えて分解し,硫酸 (1+1) 5mlを加え,蒸発して白煙を発生させる。
放冷した後,水20mlを加えて加熱して塩類を溶解する。室温まで冷却した後,溶液を1 000mlの全量
フラスコに水を用いて移し入れ,水で標線まで薄めて原液 (100μgCo/ml) とする。この原液を使用の
都度,必要量だけ水で正確に10倍に薄めて標準コバルト溶液とする。
5.3
器具 器具は,次による。
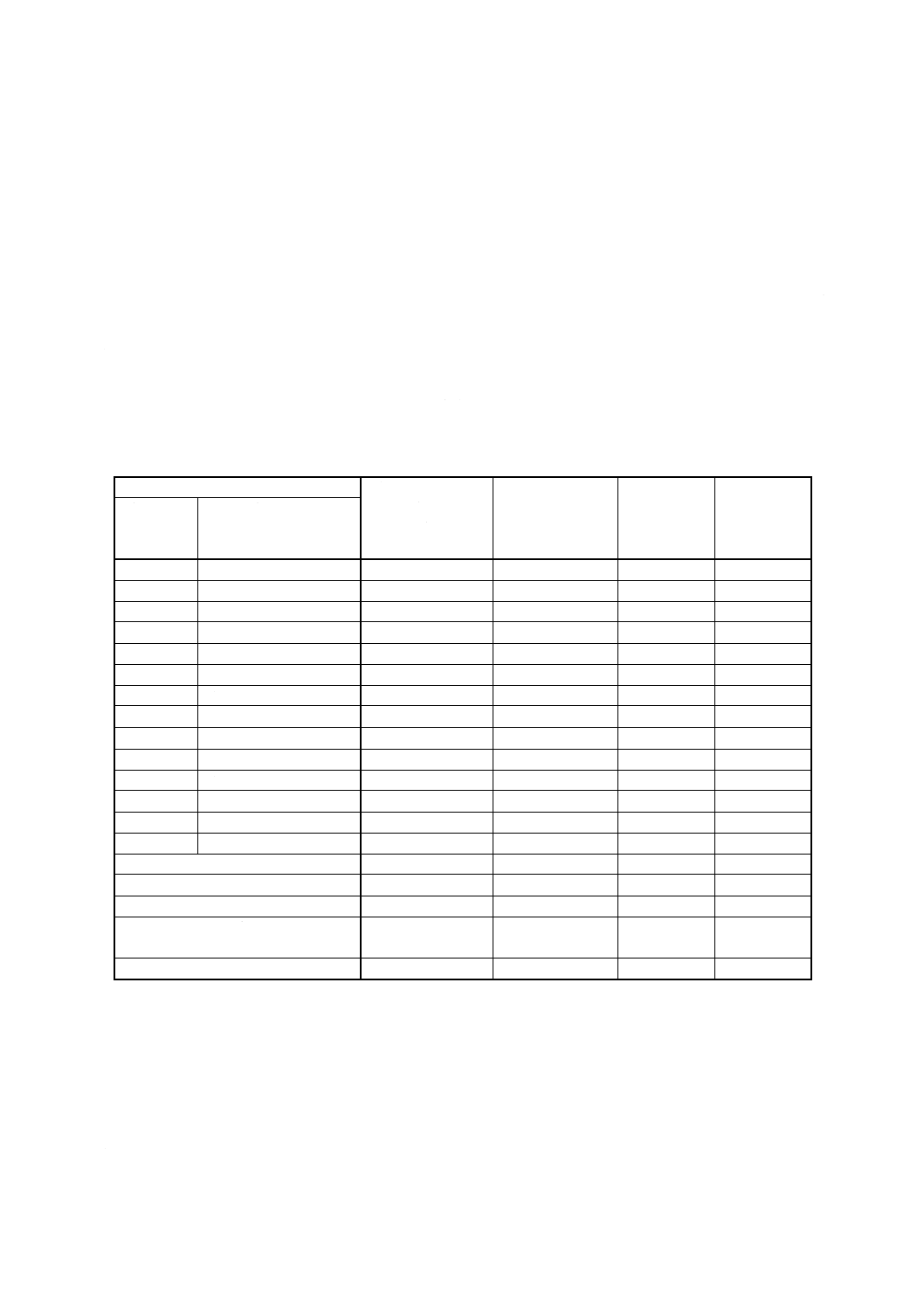
a) 円筒状白金電極 通常,図1のものを用いる。
b) らせん状白金電極 通常,図2のものを用いる。
c) 半円形時計皿 通常,図3のものを用いる。
図1 円筒状白金陰極
図2 らせん状白金
陽極
図3 半円形時計皿
5.4
試料はかり取り量 試料はかり取り量は,通常,0.50gとする。
5.5
操作
5.5.1
試料の分解 試料をはかり取って電解用ビーカー (300ml) に移し入れ,時計皿で覆い,混酸20ml
を加え,穏やかに加熱して完全に分解した後,数分間煮沸して窒素酸化物を追い出す。放冷した後,時計
皿の下面及びビーカーの内壁を水で洗浄して時計皿を取り除く。
4
H 1283 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5.5.2
銅の電解分離 銅の電解分離は,次の手順によって行う。
a) 5.5.1で得た溶液に,水を加えて液量を150mlとする。
b) 円筒状白金電極 [5.3 a)] を陰極とし,らせん状白金電極 [5.3 b)] を陽極に用い,2個の半円形時計皿
[5.3 c)] で覆い,液温を15〜30℃とし,0.3〜0.4Aの電流を通じて約12時間電解する(1)。
c) 時計皿の下面,ビーカーの内壁及び電極の柄の液面上に露出した部分を水洗し,電流を通じたまま水
洗しながら両極を徐々に引き上げて取り除く。
注(1) 電解時間を短縮するために,磁気かき混ぜ器などによって電解液をかき混ぜながら,1.0〜1.4A
の電流を通じて2〜3時間電解を行ってもよい。
5.5.3
電解後の溶液の処理 電解後の溶液の処理は,次の手順によって行う。
a) 5.5.2 c)で得た溶液(2)を,250mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
b) この溶液を,コバルト量が20〜250μgになるように分取し,ビーカー (200ml) に移し入れる。
c) 溶液を加熱蒸発して乾固した後,放冷する。硫酸 (1+3) 1滴と水10mlを加え,加熱して可溶性塩類
を溶解した後,常温まで冷却する。
注(2) この溶液の代わりにJIS H 1272の5.5.3 c)で得られる電解残液を用いることができる。
5.5.4
溶液の呈色 溶液の呈色は,次の手順によって行う。
a) 5.5.3 c)で得た溶液に,酢酸ナトリウム溶液 [5.2 d)] 5mlを加える(3)。
b) ニトロソR塩溶液 [5.2 e)] を正しく5m1加え,約1分間穏やかに煮沸してコバルトを呈色させる。
c) 硝酸 (1+1) 5mlを加え,引き続き約1分間煮沸した後,直ちに常温まで冷却する。
d) 溶液を50mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
注(3) ここで,溶液のpHは5〜6となる。pHが5〜6にならない場合には,更に酢酸ナトリウム溶液 [5.2
d)] を添加する。
5.5.5
吸光度の測定 5.5.4 d)で得た溶液の一部を光度計の吸収セル (10mm) に取り,水を対照液として,
波長520nm付近の吸光度を測定する。
5.6
空試験 試料を用いないで,5.5.1及び5.5.3〜5.5.5の手順に従って試料と同じ操作を試料と並行して
行う(4)。
注(4) 5.5.3 b)で,空試験液も試料溶液と同量分取する。
5.7
検量線の作成 標準コバルト溶液 [5.2 f)] 0〜25.0ml(コバルトとして0〜250μg)を段階的に数個の
ビーカー (100ml) に取り,水で液量を約20mlとし,酢酸ナトリウム溶液 [5.2 d)] 5mlを加える(3)。以下,
5.5.4 b)〜5.5.5の手順に従って,試料と同じ操作を試料と並行して行い,得た吸光度とコバルト量との関係
線を作成し,その関係線を原点を通るように平行移動して検量線とする。
5.8
計算 5.5.5及び5.6で得た吸光度と,5.7で作成した検量線とからコバルト量を求め,試料中のコバ
ルト含有率を次の式によって算出する。
100
250
2
1
×
×
−
B
o
m
A
A
C=
ここに,
Co: 試料中のコバルト含有率 [% (m/m)]
A1: 分取した試料溶液中のコバルト検出量 (g)
A2: 分取した空試験液中のコバルト検出量 (g)
m: 試料はかり取り量 (g)
B: 試料溶液及び試験液の分取量 (ml)
5
H 1283 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6. ヘキサシアノ鉄(III)酸カリウム滴定法
6.1
要旨 試料を硝酸と塩酸との混酸で分解し,過塩素酸を加え,白煙が発生するまで加熱濃縮して,
クロムを酸化した後,二りん酸ナトリウムを加えてマンガンとの錯体を形成させる。窒素ガスを通じて溶
存している塩素及び酸素を除去した後,くえん酸アンモニウム,硫酸アンモニウム,アンモニア水及び一
定量のヘキサシアノ鉄(III)酸カリウムを加え,電位差滴定装置を用いて一定量のヘキサシアノ鉄(III)
酸カリウムを標準コバルト溶液で滴定する。
6.2
試薬 試薬は,次による。
a) 塩酸
b) 硝酸
c) 硝酸 (1+1)
d) 過塩素酸
e) りん酸
f)
混酸(硝酸1,塩酸3)
g) アンモニア水
h) ニッケル溶液 (10gNi/l) 純粋でコバルトを含まないニッケル2gをはかり取り,ビーカー (200ml) に
移し入れ,硝酸20mlを加えて分解する。穏やかに加熱し完全に分解する。常温まで冷却した後,溶
液を200mlの全量フラスコへ水を用いて移し入れ,水で標線まで薄める。
i)
鉄溶液 (5gFe/l) 純粋でコバルトを含まない鉄1gをはかり取り,ビーカー (200ml) に移し入れ,塩
酸10mlを加えて分解する。穏やかに加熱し完全に分解する。常温まで冷却した後,溶液を200mlの
全量フラスコへ水を用いて移し入れ,水で標線まで薄める。
j)
クロム溶液 (5gCr/l) ニクロム酸カリウム (JIS K 8005) 2.8gをはかり取り,ビーカー (200ml) に移
し入れ,水約50mlに溶解する。溶液を200mlの全量フラスコに水を用いて移し入れ,水で標線まで
薄める。
k) 二りん酸ナトリウム溶液 二りん酸ナトリウム十水和物200gを沸騰している水約800mlに溶解し,
熱いうちに水で液量を1 000mlとする。この溶液は,使用する直前に調製する。
l)
くえん酸アンモニウム溶液 くえん酸アンモニウム一水和物を正確に125gはかり取り水250mlに溶
解する。注意して,一定の速度でかき混ぜながら,アンモニア水170mlを加える。冷却しながら水で
液量を500mlとする。
m) 硫酸アンモニウム溶液 硫酸アンモニウム125gを250mlの水に溶解し,水で液量を500mlとする。
n) 標準コバルト溶液 (2.00mgCo/ml) コバルト[99.9% (m/m) 以上]1.000gをはかり取り,ビーカー
(300ml) に移し入れ,時計皿で覆い,硝酸 (1+1) 20mlを加え,穏やかに加熱して分解した後,煮沸し
て窒素酸化物を追い出す。放冷した後,時計皿の下面及びビーカーの内壁を水で洗って時計皿を取り
除き,溶液を500mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
o) ヘキサシアノ鉄(III)酸カリウム標準溶液 ヘキサシアノ鉄(III)酸カリウム5.6gを水250mlに溶解
し,溶液をろ紙を用いてろ過し,ろ紙を水で洗浄する。ろ液及び洗液とを500mlの全量フラスコに移
し入れ,水で標線まで薄める。使用する直前に,再びろ過した後,標定を次の手順によって行う。
1) 試料(5)を0.2〜0.4gはかり取り,ビーカー (400ml) に移し入れ,6.5.1 b)の操作を行う(6)。
2) 標準コバルト溶液 [6.2 n)] を正しく20ml加える。
3) 6.5.1 c)〜6.5.2 c)の手順に従って操作し,ヘキサシアノ鉄(III)酸カリウム標準溶液1mlのコバルト
相当量を次の式によって算出する。

6
H 1283 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2
1
1)
20
(
00
.2
V
m
V
f
+
+
×
=
ここに,
f: ヘキサシアノ鉄(III)酸カリウム標準溶液1mlのコバルト相
当量 (mg/ml)
V1: 滴定で使用した標準コバルト溶液の量 (ml)
V2: 滴定で使用したヘキサシアノ鉄(III)酸カリウム標準溶液の
量 (ml)
m1: 1)ではかり取った試料中のコバルト量 (mg)
注(5) 試料は,鉄及びクロムを含み,かつ,コバルト含有率が0.1% (m/m) 以下で既知のニッケル合金
を用いる。
(6) 注(5)の試料が入手できない場合には,ニッケル,鉄及びクロムの量がニッケル合金と同様にな
るように,ニッケル溶液 [6.2 h)] ,鉄溶液 [6.2 i)] 及びクロム溶液 [6.2 j)] を混合した後,2)
及び3)の手順に従って標定する。
6.3
装置 装置は,次による。
電位差滴定装置:指示電極に白金を用い,参照電極として銀/塩化銀電極,カロメル電極又は硫酸水銀(I)
電極を用いて電位差が測定可能なもの。
6.4
試料はかり取り量 試料はかり取り量は,表2による。
表2 試料はかり取り量
試料中のコバルト含有率
% (m/m)
試料はかり取り量
g
2以上 10未満
0.40
10以上 25以下
0.20
6.5
操作
6.5.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 試料をはかり取ってビーカー (500ml) に移し入れる。
b) 時計皿で覆い,混酸25mlを加え,穏やかに加熱して完全に分解する(7)(8)。
c) 過塩素酸8mlを加え,加熱して過塩素酸の白煙を発生させ,クロムを二クロム酸に酸化する。さらに
過塩素酸2mlを加え,2分間過塩素酸の白煙を発生させる。放冷した後,水約10mlを用いて時計皿の
下面及びビーカーの内壁を洗浄して時計皿を取り除く。
d) 熱二りん酸ナトリウム溶液 [6.2 h)] 50mlを加え,沸騰するまで加熱し,更に数分間煮沸する。
e) 室温まで放冷した後,窒素を10〜15分間通じて,溶液中の塩素及び酸素を取り除く。
注(7) 分解しにくい合金の場合には,塩酸1mlずつを加える。
(8) 合金によっては,塩酸30mlと硝酸2mlの混酸を加える。
6.5.2
滴定 滴定は,次の手順によって行う。
a) 6.5.1 e)で得た溶液に,くえん酸アンモニウム溶液 [6.2 l)] 30m1及び硫酸アンモニウム溶液 [6.2 m)]
20mlを加える。
b) ビーカーをマグネチックスターラー上に設置し,かき混ぜる。電位差滴定装置 [6.3] の指示電極及び
参照電極を溶液中に挿入し,ヘキサシアノ鉄(III)酸カリウム標準溶液で滴定し,更に過剰に3〜5m1
加える。
c) この溶液を水で約300mlに薄めた後,アンモニア水60mlを加えた後,標準コバルト溶液で滴定する。
終点付近では滴定をゆっくりと行い,電位差滴定装置の指示が急激に変化する点を終点とする(9)。
7
H 1283 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注(9) 終点を過ぎてからも少し滴定を続ける。場合によっては,滴定曲線から図上で終点を決定する。
6.6
計算 試料中のコバルト含有率は,次の式によって算出する。
100
000
1
00
.2
4
3
×
×
×
−
×
m
V
f
V
Co=
ここに,
Co: 試料中のコバルト含有率 [% (m/m)]
V3: ヘキサシアノ鉄(III)酸カリウム標準溶液の使用量 (ml)
f: ヘキサシアノ鉄(III)酸カリウム標準溶液1mlのコバルト相
当量 (mg/ml)
V4: 標準コバルト溶液の使用量 (ml)
m: 試料はかり取り量 (g)
7. 原子吸光法(A法)
7.1
要旨 試料を硝酸で分解した後,溶液を原子吸光光度計の空気・アセチレンフレーム中に噴霧し,
その吸光度を測定する。
7.2
試薬 試薬は,次による。
a) 硝酸 (1+1)
b) ニッケル 99.9% (m/m) 以上で,コバルト含有率が0.000 1% (m/m) 以下のもの。
c) 標準コバルト溶液 (100μgCo/ml) コバルト[99.9% (m/m) 以上]1.000gをはかり取ってビーカー
(300ml) に移し入れ,時計皿で覆い,硝酸 (1+1) 40mlを加え,穏やかに加熱して分解した後,煮沸し
て窒素酸化物を追い出す。常温まで冷却した後,時計皿の下面及びビーカーの内壁を水で洗って時計
皿を取り除き,溶液を1 000mlの全量フラスコに水を用いて移し入れ,硝酸 (1+1) 160mlを加え,水
で標線まで薄めて原液 (1 000μgCo/ml) とする。この原液を使用の都度,必要量だけ水で正確に10倍
に薄めて標準コバルト溶液とする。
7.3
試料はかり取り量 試料はかり取り量は,2.0gとする
7.4
操作
7.4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 試料をはかり取ってビーカー (300ml) に移し入れる。
b) 時計皿で覆い,硝酸 (1+1) 20mlを加え,穏やかに加熱して完全に分解し,引き続き加熱して窒素酸
化物を追い出す。放冷した後,時計皿の下面及びビーカーの内壁を水で洗って時計皿を取り除く。
c) 加熱してシロップ状となるまで濃縮し,常温まで冷却した後,硝酸 (1+1) 20ml及び水約100mlを加
えて塩類を溶解する。溶液を200mlの全量フラスコに水を用いて移し入れる(10)。
d) 水を加えて標線まで薄める。
注(10) この溶液のコバルト量が5 000μg以上の場合には,水で標線まで薄めた後,コバルト量が500〜
5 000μgになるように溶液を別の200mlの全量フラスコに分取する。
7.4.2
吸光度の測定 7.4.1 d)で得た溶液の一部を水を用いてゼロ点を調整した原子吸光光度計の空気・
アセチレンフレーム中に噴霧し,波長241.2nmにおける吸光度を測定する。
7.5
空試験 7.6の検量線作成操作において得られる標準コバルト溶液を添加しない溶液の吸光度を,空
試験の吸光度とする。
7.6
検量線の作成 検量線の作成は,次の手順によって行う。
a) ニッケル [7.2 b)] を2.0gずつはかり取って数個のビーカー (200ml) に移し入れ,以下,7.4.1のb)及
びc)の手順に従って操作する(11)。
8
H 1283 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) 標準コバルト溶液 [7.2 c)] 0〜50.0ml(コバルトとして0〜5 000μg)を段階的に加え,以下,7.4.1 d)
及び7.4.2の手順に従って試料と同じ操作を試料と並行して行い,得た吸光度とコバルト量との関係線
を作成し,その関係線を原点を通るように平行移動して検量線とする。
注(11) 7.4.1 d)で注(10)を適用した場合には,水で標線まで薄めた後,7.4.1 c)で分取した試料溶液と同量
ずつを200mlの全量フラスコに分取する。
7.7
計算 計算は,次のいずれかによる。
a) 7.4.1 c)で分取をしなかった場合 7.4.2及び7.5で得た吸光度と,7.6で作成した検量線とからコバル
ト量を求め,試料中のコバルト含有率を次の式によって算出する。
100
2
1
×
−
m
A
A
Co=
ここに,
Co: 試料中のコバルト含有率 [% (m/m)]
A1: 試料溶液中のコバルト検出量 (g)
A2: 空試験液中のコバルト検出量 (g)
m: 試料はかり取り量 (g)
b) 7.4.1 c)で分取をした場合 7.4.2及び7.5で得た吸光度と,7.6で作成した検量線とからコバルト量を
求め,試料中のコバルト含有率を次の式によって算出する。
100
200
4
3
×
×
−
B
o
m
A
A
C=
ここに,
Co: 試料中のコバルト含有率 [% (m/m)]
A3: 分取した試料溶液中のコバルト検出量 (g)
A4: 分取した空試験液中のコバルト検出量 (g)
m: 試料はかり取り量 (g)
B: 試料溶液及び空試験液の分取量 (ml)
8. 原子吸光法(B法)
8.1
要旨 試料を塩酸と硝酸との混酸で分解し,乾固近くまで加熱する。塩酸を加え,乾固近くまで加
熱し,塩酸及び塩化ストロンチウムを加えた後,溶液を原子吸光光度計の空気・アセチレンフレーム中に
噴霧し,その吸光度を測定する。
8.2
試薬 試薬は,次による。
a) 塩酸
b) 塩酸 (1+1)
c) 硝酸
d) 硝酸 (1+1)
e) 混酸(塩酸3,硝酸1) この混酸は,使用の都度調製する。
f)
塩化ストロンチウム溶液 塩化ストロンチウム六水和物113.5gをはかり取り,ビーカー (1l) に移し
入れ,水約400mlを加え,加熱して溶解する。常温まで冷却した後,溶液を1 000mlの全量フラスコ
に水を用いて移し入れ,水で標線まで薄める。
g) 標準コバルト溶液 (50μgCo/ml) コバルト[99.9% (m/m) 以上]1.000gをはかり取ってビーカー
(300ml) に移し入れ,時計皿で覆い,塩酸 (1+1) 30mlを加え,穏やかに加熱して分解する。常温まで
冷却した後,時計皿の下面及びビーカーの内壁を水で洗って時計皿を取り除き,溶液を1 000mlの全
量フラスコに水を用いて移し入れ,塩酸35mlを加え,水で標線まで薄めて原液 (1 000μgCo/ml) とす
る。この原液50.0mlを1 000mlの全量フラスコに取り,塩酸50mlを加え,水で標線まで薄めて標準

9
H 1283 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
コバルト溶液とする。
8.3
試料はかり取り量 試料はかり取り量は,1.0gとする。
8.4
操作
8.4.1
試料溶液の調製 試料溶液の調製は,次のいずれかの手順によって行う。
a) 試料中のコバルト含有率が0.01% (m/m) 以上0.1% (m/m) 未満の場合
1) 試料をはかり取ってビーカー (300ml) に移し入れる。
2) 時計皿で覆い,混酸 [8.2 e)] 20mlを加え,穏やかに加熱して完全に分解する(12)。放冷した後,時計
皿の下面及びビーカーの内壁を水で洗って時計皿を取り除く。
3) 塩酸25mlを加え,乾固近くまで加熱する(13)。この操作をもう一度繰り返す。
4) 放冷した後,塩酸5m1及び水20mlを加え,加熱して塩類を溶解する。
5) 常温まで冷却した後,溶液を100mlの全量フラスコに水を用いて移し入れ,塩化ストロンチウム溶
液 [8.2 f)] 4mlを加え,水で標線まで薄める(14)。
b) 試料中のコバルト含有率が0.1% (m/m) 以上4.0% (m/m) 以下の場合
1) a)の1)〜4)の手順に従って操作する。
2) 常温まで冷却した後,溶液を500mlの全量フラスコに水を用いて移し入れ,塩酸25mlを加えた後,
水で標線まで薄める(14)。
3) 溶液を表3の分取量に従って100mlの全量フラスコに分取し,表3に従って塩酸を加えた後,塩化
ストロンチウム溶液 [8.2 f)] 4mlを加え,水で標線まで薄める。
表3 分取量及び塩酸添加量
試料中のコバルト含有率
% (m/m)
分取量
ml
塩酸添加量
ml
0.1以上 0.8未満
50.0
3
0.4以上 4.0以下
10.0
5
注(12) 完全に分解しない場合には,塩酸1mlを追加する。
(13) 乾固しないように注意する。
(14) 加水分解生成物が認められた場合には,乾いたろ紙を用いてろ過するか,又は遠心分離によっ
て除去する。
8.4.2
吸光度の測定 8.4.1のa) 5)又はb) 3)で得た溶液の一部を,水を用いてゼロ点を調整した原子吸光
光度計の空気・アセチレンフレーム中に噴霧し,波長240.7nmにおける吸光度を測定する。
8.5
空試験 試薬だけを用いて,8.4.1及び8.4.2の手順に従って試料と同じ操作を試料と並行して行う
(15)。
注(15) 8.4.1 b) 3)で試料溶液を分取する場合には,空試験液も試料溶液と同量分取する。
8.6
検量線の作成 数個の100mlの全量フラスコに標準コバルト溶液 [8.2 g)] 0〜20.0ml(コバルトとし
て0〜1 000μg)を段階的に取り,塩化ストロンチウム溶 [8.2 f)] 4ml及び塩酸5mlを加え,水で標線まで
薄める。各溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の空気・アセチレンフレーム中に
噴霧し,波長240.7nmにおける吸光度を試料と並行して測定し,得た吸光度とコバルト量との関係線を作
成し,その関係線を原点を通るように平行移動して検量線とする。
8.7
計算 計算は,次のいずれかによる。
a) 試料溶液の調製を8.4.1 a)によって行った場合 8.4.2及び8.5で得た吸光度と,8.6で作成した検量線
とからコバルト量を求め,試料中のコバルト含有率を次の式によって算出する。
10
H 1283 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
100
2
1
×
−
m
A
A
Co=
ここに,
Co: 試料中のコバルト含有率 [% (m/m)]
A1: 試料溶液中のコバルト検出量 (g)
A2: 空試験液中のコバルト検出量 (g)
m: 試料はかり取り量 (g)
b) 試料溶液の調製を8.4.1 b)によって行った場合 8.4.2及び8.5で得た吸光度と,8.6で作成した検量線
とからコバルト量を求め,試料中のコバルト含有率を次の式によって算出する。
100
500
4
3
×
×
−
B
o
m
A
A
C=
ここに,
Co: 試料中のコバルト含有率 [% (m/m)]
A3: 分取した試料溶液中のコバルト検出量 (g)
A4: 分取した空試験液中のコバルト検出量 (g)
m: 試料はかり取り量 (g)
B: 試料溶液及び空試験液の分取量 (ml)
11
H 1283 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
国際規格整合化推進本委員会並びにニッケル及びニッケル合金分析方法工業標準原案作成委員会 構成表
氏名
所属
(委員長)
神 尾 彰 彦
東京工業大学工学部
後 藤 敬 一
通商産業省基礎産業局非鉄金属課
◎ 大 嶋 清 冶
工業技術院標準部材料規格課
村 田 祐 滋
東京都立工業技術センター金属部
竹 内 孝 夫
科学技術庁金属材料技術研究所
◎ 橋 本 繁 晴
財団法人日本規格協会
太 田 裕 二
社団法人日本銅センター技術部
大 屋 武 夫
ステンレス協会
佐 藤 秀 樹
社団法人日本電子材料工業会技術部
稲 垣 勝 彦
日本鉱業協会技術部
赤 峰 淳 一
社団法人日本電機工業会技術部
篠 原 脩
社団法人日本ガス石油機器工業会技術部
山 添 哲 郎
通信機械工業会技術部
村 岡 良 三
社団法人日本自動車部品工業会技術部
山 下 満 男
富士電機株式会社生産技術研究所
安 井 毅
株式会社東芝材料部品事業部開発技術部
◎ 田 中 尚 生
三菱マテリアル株式会社桶川製作所
恒 原 正 明
古河電気工業株式会社金属事業本部
菅 沼 輝 夫
日鉱金属株式会社倉見工場技術部
大 関 哲 雄
大木伸銅工業株式会社技術部
中 島 安 啓
株式会社神戸製鋼所アルミ・銅事業本部技術部
田部井 和 彦
三菱マテリアル株式会社桶川製作所
岡 村 明 人
三菱伸銅株式会社若松製作所品質保証部
◎ 藤 沢 裕
日本伸銅協会技術部
○ 町 田 克 巳
住友金属鉱山株式会社中央研究所分析センター
○ 山 下 務
株式会社東芝材料部品事業部品質保証部
○ 久留須 一 彦
古河電気工業株式会社横浜研究所分析技術センター
○ 中 村 靖
株式会社ジャパンエナジー分析センター顧問
○ 豊 嶋 雅 康
住友軽金属工業株式会社研究開発センター
(関係者)
和 田 隆 光
財団法人日本規格協会技術部国際整合化規格室
相 馬 南海雄
日本伸銅協会総務部
山 本 寿 美
古河電気工業株式会社横浜研究所
天 川 義 勝
株式会社ジャパンエナジー分析センター
備考 ◎印:本委員会及び原案作成分科会委員
○印:原案作成分科会委員